Dr. Naikwadi College of Pharmacy Jamgaon, Sinnar, Nashik, 422103
The affinity chromatography method of separating biochemical mixture based on a very particular interaction between the antigen and a protein, an enzyme, a substrate, a receptor, and ligand, or nucleic acid and protein. Additionally, its affinity purification and preparation of solutes as a result, chromatography also possesses a potential for investigating the functional roles of purified the reactants there. Purification of affections can save a lot of time and help with several 100 times or more purification, but success depends on the approach taken. This review explains about the fundamental idea, procedure, and applications.
Bioaffinity in combination with Affinity was developed through chromatography. chromatography (no. 1). Affinity chromatography is a kind of liquid chromatography that uses biological-like interactions for the separation and specific analysis of sample components. The technique has high selectivity, which results in high resolution and typically high protein capacity of significance. Purification can be in the order of several thousand times, and active recovery material are frequently extremely high. Affinity chromatography makes use of particular binding interactions among the relevant analytes.and a binding partner or ligand (immobilized on the phase stationary). A common type of affinity chromatography procedure, a solid is attached to the ligand. matrix that is insoluble, typically a polymer like as a chemically modified polyacrylamide or agarose to introduce functional groups that are reactive, the ligand can react, forming stable covalent bonds(4)
This particular method is known as the most precise and efficient method for protein purification. The biomolecules' separation is based on extremely particular biological interactions between two different molecules, like an enzyme and the substrate. Affinity chromatography (also called affinity purification) utilizes particular binding interactions among atoms and molecules An individual ligand is chemically bonded to a solid or immobilized. support so that a complex mixture can be passed through across the column, those molecules having specific.
The ligand's binding affinity is bound. After The remainder of the sample components are washed away, The support is removed from the bound molecule, as a result, it was separated from the original sample. The fundamental concept has been extensively utilized as a powerful instrument for separating and purifying a wide range of macromolecules used in biology. Its Selectivity determines purification's effectiveness. of interaction, and thus of adsorption, of a an affinity adsorbent-bound biological macromolecule which is prepared by the covalent immobilization a particular ligand on a solid polymeric matrix.The use of affinity chromatography is not necessary. the charge, hydrophobicity, or molecular weight additional physical characteristics of the relevant analyte to be known, despite the fact that knowing its binding properties is useful in the design of a separation protocol.
Table No.1: Interaction of Typical Biological Which is Used in Affinity Chromatography
|
Sr. No |
Types of Ligands |
Target Molecule |
|
1 |
Substrate analogue |
Enzymes |
|
2 |
Antibody |
Antigen |
|
3 |
Lectin |
Polysaccharides |
|
4 |
Harmone |
Receptor |
2. HISTORY
Emil, a German scientist, died in 1910. An article written by Starkenstein described the idea of solving complex macromolecules through their interactions with a person who is stuck substrate. The influence was discussed in this manuscript. of chloride on the liver's enzymatic activity - amylase and opened the door for the early beginnings of this strategy by a number of researchers.
The term affinity chromatography introduced in 1968 by Pedro Cuatecasas, Chris In an article, Anfinsen and Meir Wilchek briefly described the enzyme method purification via immobilized substrates and inhibitors. Other early articles described the Cyanogen bromide–mediated activation of the Sepharose matrix the reaction of bromide (CNBr) and the use of a spacer arm to eliminate steric obstruction. Since 1968,
The field of affinity chromatography has established itself as a common method for enzyme separation in the laboratory. Utilization of affinity chromatography for determination of the constants of inhibition Enzymes appear to have bright futures. On the the basis for the enzyme's elution volumes using the immobilized inhibitor from the column a variety of soluble inhibitor concentrations—the With and without, inhibition constants can be determined. with soluble inhibitors and bound inhibitors employed.(10)
FUNDAMENTAL PRINCIPLES OF CHROMATOGRAPHY OF THE AFINITY:
Utilizing a desired protein for separation affinity chromatography relies on the reversible interactions between the purified protein and the chromatographically coupled affinity ligand matrix. As previously stated, the majority of proteins have a site with built-in recognition that can be used to select the appropriate affinity ligand. The securing between the selected protein and the protein of interest ligand must be both specific and reversible
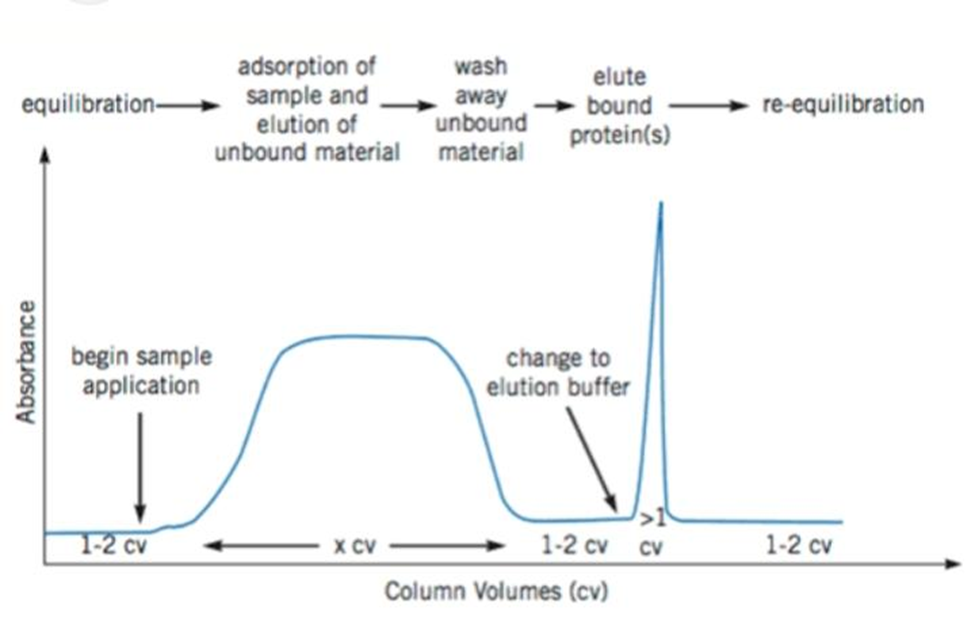
Figure.1 Typical Affinity Chromatography Purification
Batch Chromatography vs Column Chromatography :
The solid phase binding may be accomplished through column chromatography.The first column is filled with solid medium. mixture that goes through the column to make settling possible, a wash buffer run through the column and the elution buffer was then put on the column and collected.Most of the time, these steps are done in ambient pressure. On the other hand, binding can be achieved. utilizing a batch treatment, such as by incorporating the initial mixture to the solid phase in a vessel, blending, separating the solid phase, and getting rid of the washing in the liquid phase, centrifuging again, and adding the elution buffer, re-centrifuging and removing the elute.Sometimes a hybrid method is employed so that the batch method is used for binding, however, the solid phase containing the bound target molecule is packed onto a column and washing and elution are carried out on the column.
ligands utilized in affinity chromatography comes from both organic as well as non-organic sources. Illustrations of biological Serum proteins, lectins, and antibodies are sources. Metal chelates, moronic acts, and other inorganic sources and dyes with triazine. A third approach, increased bed absorption, which combines the advantages of the two methods referred to previously, has also been developed. The Particles of the solid phase are arranged in a column at The liquid phase enters from the bottom and exits at the top. The particles of gravity makes sure that the liquid phase does not follow the solid phase out of the column.
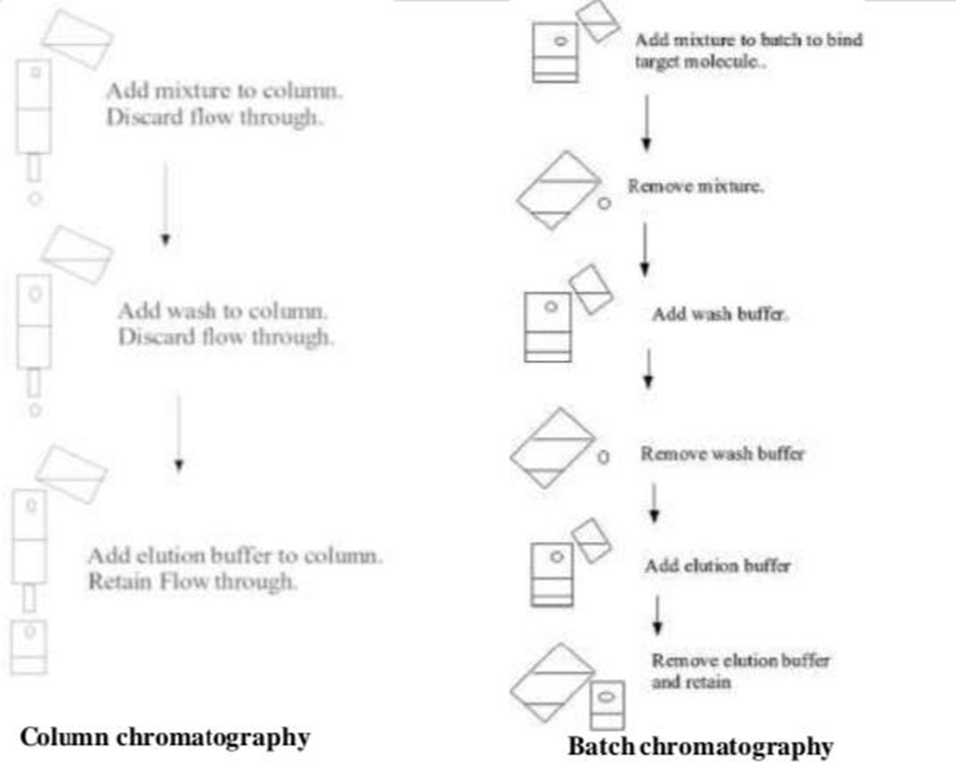
Figure 2. Batch chromatography Vs Column chromatography
TYPES OF AFFLUENCE CHROMATOGRAPHY:-
Affinity :methods that use boronic acid or boronates as ligands are one group of Chromatographic techniques that have been used successfully with clinical samples. This group of methods, collectively referred to as "boronate affinity" chromatography includes one of the earliest reported the quantitative use of affinity chromatography in the clinical laboratory, specifically, the determination of glycohemoglobin for the assessment of long-term diabetes management at a pH above 8, most boronate derivatives form covalent bonds with compounds that are composed of cis-diol groups structure. Because sugars such as glucose possess cis-diol groups, boronates are valuable for resolving glycoproteins from non-glycoprotein.
The class of ligands known as lectins have been used for the direct detection of clinical analytes by affinity chromatography. The lectins are non-immune system proteins that have the ability to recognize and bind certain types of leftover carbohydrates Lectin affinity's clinical application chromatography has been in the separation and examination of isoenzymes. where an HPLC column containing immobilized wheat germ agglutinin was used to differentiate between bone-derived and liver-derived alkaline phosphatase isoenzymes in human serum.
A third class of ligands that have been used in direct analyte detection by affinity chromatography are antibody-binding proteins such as protein A and protein G. These are cells of bacteria. Wall proteins produced by Staphylococcus aureus and group G streptococci, respectively. The ability to bind to these ligands the constant region of many types of immunoglobulins. Protein A and protein G bind most strongly to immunoglobulins at or near neutral pH, but are easy to separate from when placed in a buffer with a pH that is lower, solutes. Protein A and protein G's capacity to These are excellent ligands for the based on immobilized protein A for the analysis of because they bind to antibodies. IgG in serum samples
Each and every kind of affinity chromatography, those that use antibodies or antibody fragments as, the largest and most diverse group is made up of ligands. of clinical testing affinity methods. This is a combined result of the specificity of antibodies and the relative ease with which they can be obtained to a wide variety of analytes. The immunoaffinity chromatography? (IAC) is method used for an method of affinity chromatographic. and in which the stationary phase consists of an antibody or antibody-related reagent. When such a technique is performed as part of an HPLC system, the resulting method can be referred to as ?high-performance immunoaffinity chromatography.
AFFINITY PURIFICATION:-
A technique called affinity chromatography selective purification of a molecule or group of molecules from complex mixtures based on highly specific biological interaction between the two molecules. Typically, the interaction can be reversed. and purification is achieved through a biphasic interaction with one of the molecules (the ligand) immobilized to a surface while its partner (the target) is in a mobile phase as part of a complex mixture. These step of capture is generally followed by elution and washing, resulting in recovery of highly purified protein. Interactions that are highly selective allow for a fast, often single step, process, with potential for purification in the hundreds to the order of thousand-fold.
Affinity chromatography can be used in a number of uses, including that of nucleic acid purification, protein purification from cell free cans, blood extracts, and blood purification provide significant time savings and several hundred-fold or higher purification, but success depends on the method used. As a result, it is crucial. to make the purification procedure work best so that efficient capture and maximum recovery of the target. By using affinity chromatography, one can separate proteins that bind to a certain fragment from proteins that do not bind that specific fragment.Because this technique of purification relies on the biological properties of the protein needed, it is a useful technique and proteins can be filtered multiple folds in a single step.
Steps Involed in Purification:
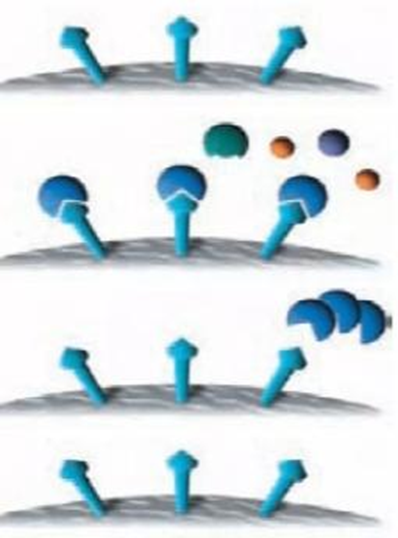
1 Step: First an immunoadsorbent is made. This is made up of a solid matrix to which the antigen is attached. Agarose, sephadex, derivatives of Polymers like cellulose can be used as the matrix.
2 Step: The serum is applied to the immunoadsorbent. As long as the capacity of the column isn't overflown, and those antibodies in the mixture specific for the antigen (shown in red) will bind (noncovalently) and be retained. The antibodies of other specificities (green) and other serum proteins (yellow) will pass through unimpeded
3 Step : A reagent is passed into the column to release the antibodies from the immunoadsorbent. buffers that have a lot of salt in them and/or low pH are often used to disrupt the noncovalent interactions between antibodies and antigen. A denaturing agent, such as 8M urea, will also break the interaction by altering the configuration of the site where the antigen binds molecule of the antibody
4 Step : Affinity medium is re-equilibrated with binding buffer. After that, the eluate is dialyzed against, for example, buffered saline in order to remove the reagent used for elution.
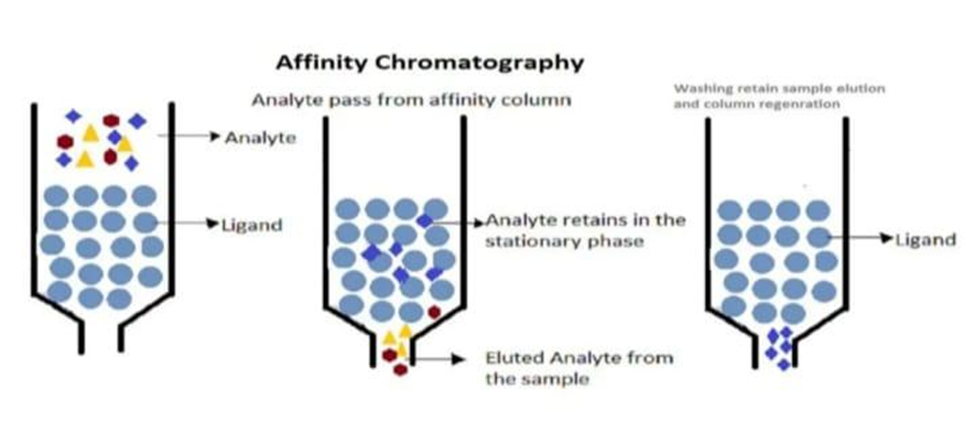
Fig.3 : Steps for purification of affinity chromatography
SEPARATION PROCEDURE IN CHROMATOGRAPHY OF AFINITY:
A sample that contains the compound In the mobile phase of an affinity chromatography, interest is focused on the affinity column. While the sample is passing through the column compounds that work in conjunction with the affinity ligand will bind. However other solutes in the sample will typically be eluted or washed off. from the column a compounds that were not retained. After all non-retained components are washed off the column, binding solute or together with ligand as solute-ligand complex are eluted by presence of mobile phase which was prepared in suitable pH, ionic strength and solvent composition for solute-ligand binding. This solution that is referred as the application buffer present the weak applying a solvent. This solvent which is referred as elution buffer represents the strong mobile phase regarding the column. Later, all of the substances of interest are eluted from the column, then application buffer is applied and the column is allowed to regenerate prior to the subsequent application sample.
A procedure for separation in affinity chromatography can be easily depicted in the manner shown in Fig.4
The content apart from the analyte passes through the column without or with weak binding to the ligand while the analyte is retarded. Following the analyte is obtained generally by using an elution in order to prepare the buffer, the column is regenerated by washing it with the application buffer. column for the next injection.
In the Figure. 5, a typical scheme of an affinity The use of chromatography is demonstrated.

CURRENT TECHNIQUES INCLUDING FINITY CHROMATOGRAPHY
Currently, affinity chromatography being used for a lot of different things ranging from the study drug protein binding interactions to the depletion of high abundance proteins to enhance the detection/quantification of dilute proteins. Using affinity chromatography, used to study drug-protein binding interactions.
Drug protein binding constants and kinetics can be measured using frontal analysis, zonal elution, and the Hummel Dreyer method. to quantify the various interactions' properties to determine drug binding and to identify allosteric interactions sites.
Two types of drug-protein binding constants exist: review articles. Information on quantifying kinetic properties of drug-protein interactions can be found in a review. A discussion on the quantification of by means of affinity chromatography, allosteric interactions can be seen in an article. Additional information on the identification of drug-binding sites can be found in a review article. When trying to analyze low abundance proteins, it is often necessary to prior to analysis, get rid of proteins that are abundant. Low abundance is effectively enriched by this removal. proteins and allows more of them to be identified and measured. Removal of the top 7 or top 14 high-abundance proteins has been shown to result in a 25% increase in identified proteins. Moreover, affinity chromatography is widely used in many omics‘ studies (e.g. proteomics, metabolomics and genomics) and is currently utilized alongside other methods to develop high-throughput methods for screening potential drugs.
USE IN APPLICATIONS OF FINITY CHEMICAL ANALYSIS:
Both can bind and immobilize antibodies. covalent and adsorption methods. Random covalent immobilization methods generally link antibodies to the solid foundation through their own free Nhydroxysuccinimide, cyanogen bromide, and amine groups N,N‘-carbonyldiimidazole, either tosyl or tresyl chloride Alternatively, an amine groups is able to react with either an aldehyde or a free epoxy group present on the activated support. As these are random immobilization methods, the antibody binding sites may be blocked due to improper orientation, multi-site attachment or steric hindrance. Site-specific covalent immobilization of antibodies can be achieved by converting the leftover carbohydrate located in the Fc region of the antibody to produce aldehyde residues which can react with amine or hydrazide supports. Covalent chemistry specific to the location The carbohydrate residues in antibodies can be converted into immobilization agents. Aldehyde production from the antibody's Fc region residues that can react with hydrazide or amine supports. Another site-specific immobilization of antibodies can be accomplished by utilizing the free sulfhydryl groups of Fab fragments. These The antibody can be coupled with groups. fragments to an affinity support using a variety of established methods including epoxy, divinylsulfone, iodoacetyl, bromoacetyl, thiol, TNB-thiol, maleimide, tresyl chloride, or tosyl chloride methods.
Antibodies can also be immobilized by adsorbing them onto secondary ligands. For example, if an antibody is reacted with hydrazide.The hydrazide biotin can react with oxidized carbohydrate residues on the Fc region of the antibody. The resultant biotinylated antibody can then adsorbed onto streptavidin or avidin affinity assistance This type of biotin immobilization allows the immobilization of the at-a-glance antibody and can be carried out with commercial equipment available biotinylationkits. Antibodies, on the other hand, can be directly adhesion to a protein A or G support due to the specific interaction of antibodies with protein A and G. Immobilized antibodies on the protein A or G support can easily be replaced by using a strong eluent, regenerating the protein A/G, and reapplying fresh antibodies. In most cases, this approach is used when a support with high capacity and high activity is needed. If a more permanent immobilization is desired, the adsorbed antibodies may be crosslinked to the support material using carbodiimide or dimethyl pimelimidate.
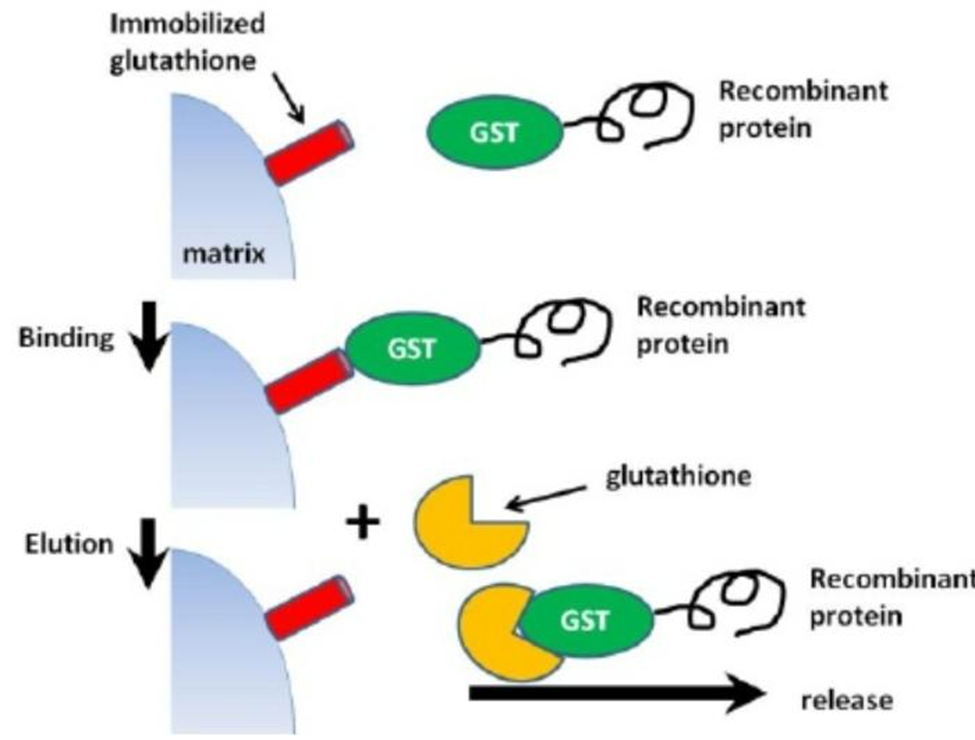
Purification of proteins can be easier and simpler,if the protein of interest has a known sequence tag.this tag may vary from a few amino acid to whole domains or even entire proteins, and it function as marker for the protein.expression and to helpfacilitate protein purification. In general, the most commonly used tags are glutathione-S-transferase (GST), histidine fusion (His or polyHis tag) and protein A fusion tags other types of fusion tags are also available including maltose-binding protein, thioredoxin, NusA, GB1 domain for protein G, and others. The decision to use any of these tagging methods depends mainly on the researcher's requirements.
Because of the GST tag, it is possible to separate and purify proteins that are GST-tagged. is able to bind glutathione, its substrate, (Glu-Cys-Gly tripeptide). Whenever glutathione reduced (GSH), it can be immobilized onto a solid support through its sulfhydryl group. Using this property, glutathione can be cross linked to an agarose bed, which then captures GST or GST tagged protien via their substrate binding interaction. Binding is most efficient near neutral physiological conditions (pH 7.5) using Tris saline buffer and mild conditions to preserve the structure and enzymatic function of GST. because of the possibility of permanent denaturation, denaturing elution conditions are not compatible with GST purification. Additionally, during denaturation or reduction, the GST fusion tag's structure frequently degrades. Following a step of washing to get rid of unbound samples, the bound GST-fusion protein can be recovered by the addition of excess reduced glutathione since, The affinity of GST for free glutathinone exceed its affinity for glutathione in the immobilised form. The free glutathione replaces the glutathione that is stuck and makes it available the GST-tagged protein from the matrix allowing its elution from the column.
Recombinant proteins which have Histidine tags can be purified using immobilised metal-ion Chromatography (IMAC),and these tags can be placed at either the N-or C-terminus of the protein.Optimal binding and, therefore, purification efficiency is achieved when the His-tag is freely accessible to metal ion suppor. Histidine tags have strong affinity for metal ions. (e.g. Co2+, Ni2+, Cu2+, and Zn2+). One of the initial materials used to support immobilized iminodiacetic acid which can bind metal ions and allow for the coordination complex with the history tagged protein.One drawback of iminodiacetic acid fosters the possibility of metal ion leaching. leading to a decreased protein yield. Modern materials for support, such as nickel-nitrilotriacetic cobalt-carboxymethylasparate and acid (Ni-NTA) (Co-CMA), which demonstrate little leaching and, as a result, result in protein purifications that are more effective.
The His-tag offers the advantages of small affinity ligand,and this limited size enusers that it has little influence on the proteins folding. In addition, if the his tag placed on the end terminal of the protein it can easily be removed using a protoprotease Polyhistidine tags are another benefit of using Histag purification methods. It is capable of binding protein in both native and denaturing conditions.The use of denaturing condition of becomes important when proteins are found in inclusion bodies and must be denatured so they can be solubilized.
Disadvantages of using His-tag protein
Potential Histag degradation, the formation of dimers and tetramers, and coelution are purification steps. other histidine-containing proteins. First, when a few Histidine residues undergo proteolytic degradation, and the The tagged protein loses a lot of its affinity. resulting in a decrease in the yield of protein. Second, once a protein has a His-tag added to its structure, it has the potential to form dimers and tetramers in the presence of ions of metal. While this usually does not pose a significant problem,it may lead to an incorrect estimation of the tagged proteins molecular mass.A further drawback of his-tag-based purification is the co-education of endogenous protein that naturally possess two or more adjacent histidine residues.
A, G, and L are either native or microbial-derived recombinant proteins that specifically bind to immunoglobulins, such as IgG, or immunoglobulin G Eighty percent is IgG. of immunoglobulins in the serum. The most well-liked support structures or matrixes for affinity applications which makes use of protein A, G, or L is agarose beads. In, clones of native and recombinant protein A Aureus staphylococcus Protein G recombinant (cell surface protein) Streptococcus has a clone of it. while the clone of recombinant protein L comes from Magnus peptidostreptococcus Both G and A protein specifically bind to IgG's Fc region, whereas protein IgG's kappa light chains are bound by L. The most widely used supports or matrixes for applications of affinity that make use of protein A, G, or L is agarose with beads (e.g. CL-4B sepharose; agarose with 2,3-dibromopropanol crosslinked and under reductive conditions, desulphated by alkaline hydrolysis conditions), polyacrylamide, and magnetic beads.All three proteins bind extensively with the IgG subclass.The journal reports that proteins A is better suited for isolating IgG from cat,dog,rabbit and pig while Protein G is typically preferred for purify IgG from use or human sample. A combination of protein A and G is also applicable for purifying a wide range of mammalian IgG samples. Since protein L binds to the kappa light chain of immunoglobulins and these light chains exist in other immunoglobulins (i.e IgG, IgM, IgA, and IgE), protein L is suitable for the purification of different classes of antibodies. A summary of the binding properties of antibody binding protein A,G and L to various immunoglobulin is provided in Table 2. IgGs from most species bind to protein A and G near physiological pH and ionic strength. To elute purified immunoglobulins from protein G sepharose, the pH should be less than 2.7. Based on total IgG binding, L, binding affinity proteins are able to bind to Kappa light chains, whereas The FC region is bound by A and G.
If it is possible to incorporate a biotin tag into a biomolecule, a streptavidin or avidin affinity method can be used to purify the biomolecule. support. Inserting a biotinylation is one option. sequence into a protein that is recombinant. Biotin protein Biotin can then be added with ligase during a step of translational modification. Biotin, also known as vitamin B7 or H, is a relatively insignificant cofactor that is found in cells. In affinity chromatography it is often used an affinity tag due to the strong interactions it has with avidin and streptavidin. A benefit of using biotin as an The affinity tag is that it has little effect on due to its small size, the activity of a large biomolecule (244Da).Streptavidin is a large protein (60 kDa) that can be obtained from Streptomyces avidinii and have an affinity constant of 1013 M–1 for biotin. Glycoprotein avidin is slightly larger (66 kDa). with slightly stronger biotin binding (1015 M–). 1). Streptavidin and avidin both have four subunits. that are each capable of binding one biotin molecule. To purify streptavidin, a biotinylated biomolecule, is used to immobilize on a support material get the molecules that have been biotinylated out of the solution. Immobilization of avidin and streptavidin is possible. amine- reactive used in a coupling chemistry. In addition, avidin can also be immobilized via its residues of carbohydrates. Biotin and due to their strong interaction (strept) avidin require severe elution conditions. to tear the binding loose. For example, 6 M guanidine hydrochloride at pH 1.5 is commonly used to elute the biomolecule with biotinylation bound. This obstructs the majority of proteins returning to their active state. To overcome this difficulty, modified (strept)avidin or a lower can be made by using biotin that has been modified. affinity interplay In one study chemically The modified avidin binds fairly well. (>109 M–1), but was also able to completely at a pH of 10, let biotinylated molecules out. In addition, at any pH between 4 and 10, a 0.6 mM biotin solution could be used to displace and elute the biotinylated molecules.
The used of biotin in a isotopically coded affinity tags (ICATs) which can be used to examine the protein content of two distinct sample. There are two labels on the ICAT: one which has heavy deuterium in it and one only consists of hydrogen (light). The two labels (light and heavy) are added to the cell separately. lysates being compared. Because the reagent includes a thiol-specific reactive group, it will covalently bind proteins' free cysteines. The lysates with labels are combined, digested with trypsin, and then isolated on a streptavidin column. Within a second separation step, the labelled proteins are analyzed using mass spectrometry. The change in protein. Then, expression between the two lysates of cells can be evaluated.pertaining to the various conditions applied to both sets of lysates of cells.
Purification of affinity is a useful tool for removing and cleaning up the excess albumin and macroglobulin contamination from samples since these components may obscure or hinder subsequent analysis steps (like mass spectrometry) as well as immunoprecipitation One method of purification which can be used to get rid of these pollutants either before or after additional steps of purification is A affinity chromatography using blue sepharose. This way method, the dye ligand is linked to by a covalent bond. sepharose through a ring of chlorotriazine. Albumin keeps non-specifically through electrostatic and/or interactions that are hydrophobic with the aromatic anionic ligand. Cibacron is the most commonly used dye. blue F-3-GA which can be immobilized onto sepharose in order to make an affinity column. This color is capable of removing more than 90% of the albumin from sample .
There are many areas connected to affinity chromatography, which has also included great interest in pharmaceutical and biomedical analysis. The utilization of is one such area. affinity columns for the isolation of enzymes. This approach may make use of affinity ligands that proteins, substrates, inhibitors, or cofactors that are connected to the biochemical routes of the goal enzyme.
One of the methods is lectin affinity chromatography. the most effective methods of learning glycosylation as a protein post translational modification. Lectins bind to carbohydrates. proteins that contain two or more carbohydrate binding sites and fall into one of five categories. based on how specific they are to the monosaccharide. They have the strongest affinities. for mannose, fucose, N-acetylneuraminic, galactose/N- acetyl galactosamine, and acetylglucosamine acid. Protein is bind to using this affinity method. a lectin that has been immobilized by its sugar moeities (N-linked or O-linked). After the glycosylation, the affinity support, the protein is bound to unbound contaminants are washed away, and the Eluted protein was purified. Numerous lectins are currently available for sale in available in a form that cannot be moved. One of them Sepharose, Concanavaline A (ConA), and wheat WGA, or germ agglutinin, are the most widely used for purification of glycoproteins. Preparing lectin affinity columns is possible. through the immobilization of lectins with various specificities toward a variety of matrices containing oligosaccharides, including monolithic stationary, silica, and agarose phases and cellulose. These lectins that have been stuck are invaluable instruments for separating and isolating glycolipids, polysaccharides, and glycoproteins cells and subcellular particles Additionally, lectin affinity columns can be used to purify detergent solubilized cell membrane components. In addition, are useful for determining whether levels have changed or during the formation of a cell, the composition of the surface glycoproteins. and in malignant or virally modified variants In the following chapters, more detailed examples of lectin affinity purification can befound.
Affinity chromatography has a wide range of applications for protein purification, such as immunoaffinity, purification of immunoglobulins, glycoprotein purification, Proteins that bind to DNA, receptor proteins, enzymes, isolation of cells, and isolation of nucleotides.
A specific purification technique is affinity chromatography. technology based on how the body works or specific chemical structure The implementation of This method involves the separation of active biomolecules that are functionally or denaturally different forms when pure proteins are isolated present at low concentrations and utilized for the removal particular contaminants.
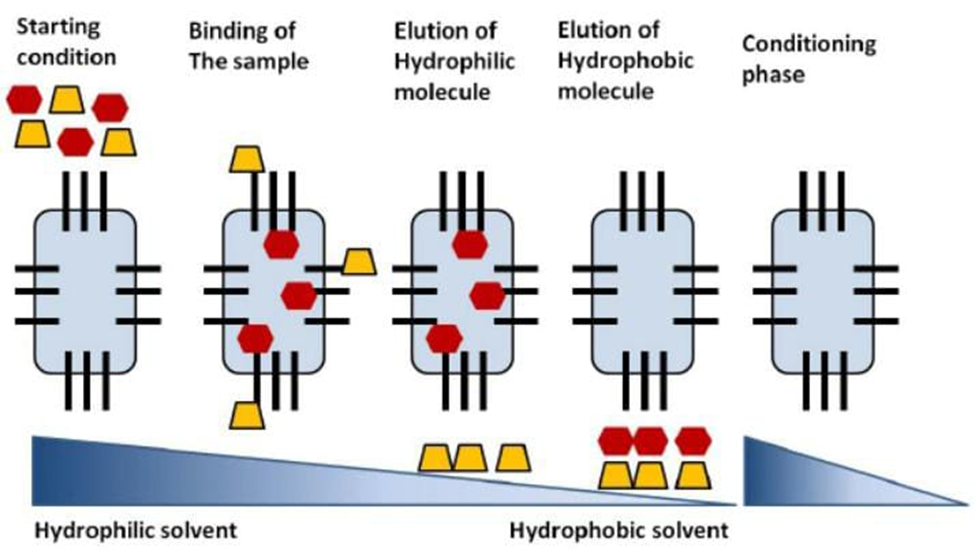
Reversed phase chromatography is a kind of an affinity relationship between biomolecules dispersed in a solvent (the mobile phase) that contains some hydrophobicity (e.g. proteins, peptides, and nucleic acids) and a hydrophobic ligand that is stuck in place (stationary phase). Chromatography with a reverse phase is generally more suitable for separating nonvolatile molecules. The expression "reversed phase" was adopted because the relationship between two molecules and a hydrophobic ligand (octadecyl; C18) in a polar aqueous phase which is reversed from normal phase chromatography is used [when a hydrophilic A hydrophobic polar ligand binds to molecules. mobile phase that is not polar]. The macromolecules (for example, peptides or proteins) are adsorbed onto column's hydrophobic surface. Elimination is achieved by means of a mobile phase, typically combination of water and organic solvents (such as acetonitrile or methanol) are sprayed onto the column in the form of gradient, such as 95:5 aqueous: organic, for example and gradually increasing the organic phase until the elution buffer is 5:95 organic: aqueous). The hydrophobic surface of is bound by macromolecules. remain in the column until the concentration of the organic phase is high enough to elute the macromolecules from the hydrophobic surface. Utilizing the reversed phase the most polar macromolecules, chromatography are eluted first and the most nonpolar.
The macromolecules that are the most polar are eluted last. The more (hydrophilic) a solute is, the quicker it elutes and vice versa. In this summary, separations of chromatography reverse phase depend on the reversible adsorption/desorption of solute molecules with a variety of hydrophobicities toward a stationary hydrophobic phase. As illustrated in Figure 12, the initial step Equilibration is a necessary step in reversed phase separation column under suitable conditions such as a (pH, ionic strength and polarity). The solvent's polar orientation can be modified by adding a solvent such as acetonitrile or methanol and an ion-pairing agent such as trifluoroacetic acid or formic acid may be added. The sample is then applied and bonded to the matrix that is immobile. Following this procedure, desorption and elution of the biomolecules is achieved by lowering the mobile's polarity phase (by boosting the proportion of organic change during the mobile phase). At the conclusion of the separation, the mobile phase should be nearly 100% natural to guarantee complete elimination of all bound substances. After everything has disappeared from the column, where the initial mobile phase is used again to the column to re-equilibrate the column for a subsequent sample application.
One is provided by affinity chromatography. important technique for determining and characterizing the intermolecular interactions.
When interaction of the initiating protein. Typically, chromatography involves linking one transferring the protein to an insoluble matrix and incubating that matrix with a solution containing possible partners who are bound. This solution could be as simple as homogenous solution containing a single recombinant protein.or intricate, after exposing the substrate protein that has been immobilized to its potential partners in binding and washing away information that is not specifically related to the The eluted and detected binding partners are any variety of methods from chromatographic detection.
Japan's JSPS supported this work. Grant from the Society for the Promotion of Science for scientific inquiry to Dr. SamehMagdeldin (23790933) from the Ministry of Culture, Japan's sports, science, and technology. The Funders had no say in whether or not to publish. the process of creating these chapters
REFERENCES


Aarti Nanne, M. S. Khatake, Gayatri Jadhav, A Review on Affinity Chromatography, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 1501-1515. https://doi.org/10.5281/zenodo.17857761
 10.5281/zenodo.17857761
10.5281/zenodo.17857761
