Department Pharmaceutical Technology. LBYP College Of Pharmacy Pathri India
This paper proposes a comprehensive, multidisciplinary method to developing a medication with an acceptable safety profile. The key parameters to examine for drug safety evaluation based on this comprehensive methodology in clued the following :1Pharmacology: Toxicity May result from drug-target interactions, such as interactions with unwanted molecular targets or molecular targets inunintendedorgans2. Chemistry: Chemicals caff old in gand side-chains pose safety risks. Toxicology3: In vivo toxicity in animals, as well as in cultured animal and human cells.1 Drug metabolism and pharmacokinetics: Safety issues related to toxification or detoxification, organdistribution, clearance, and pharmacokinetic drug-drug interactions.1riskfactorsare physiological, environmental, and genetic elements that may increase a patient us ceptibility. Anin targeted, multidisciplinary evaluation is offered.
"Everything contains poison, and nothing is free of it. Only the dose decides whether something is or is not poisonous." This toxicological theory, developed in the 15th century, serves as the foundation for modern toxicology practice. The dose-response relationship is the most relevant data set for determining safety. Drug safety is calculated using the Therapeutic index, which is a ratio the harm do set other dose required for efficacy. Because of Paracelsus' Principle, toxicologists generally assume that safety can be asses se dusing dose-response relationships without the necessity for mechanistic characterization.
FIG.1.Metabolism, And Environmental and Genetic Risk Factors
This empirical approach to safety evaluation is apparently nota de quate, judging from the
Number of drugs with serious, sometimes fatal adverse effects, which have been erroneously concluded to have an acceptable toxicity profile in preclinical and clinical safety studies. It is proposed here that drug toxicity should be defined based not only on dose– response relationship, but also as a function of pharmacology, chemistry, metabolism, and environmental and genetic risk factors.
Comprehensive approach to drug safety evaluation
The proposed comprehensive approach in drug safety evaluation is based on an integrated, multidisciplinary approach. This comprehensive understanding of drug safety should be applied towards all phases of drug discovery and development, from target identification through clinical trials.
FIG.2.Drug Metabolism and Toxicology
The key scientific disciplines to be included in this comprehensive approach to drug safety evaluation include pharmacology, chemistry, drug metabolism and toxicology. A new discipline of risk factor identification is also proposed.4
Understanding the safety concerns related to the intended pharmacological effects of drugs is logical, considering they are designed to be pharmacologically active. The toxic consequences of the drug candidate interacting with the desired target in both target and non target tissues, and the probability of interacting with unintended targets, need to be clearly established5. This is particularly crucial for a new target with limited existing clinical information. For example, a new target in the cell signaling pathways, which could serve a variety of cellular roles. Either an tag on zing or agonizing molecular target in order to treat a disease or reduce symptoms could result in negative side effects because of the impact on the target's normal functions and the chain reaction of events triggered by interaction with the molecular pharmacology target, ultimately causing or gandamage6. Anti cancer medications demonstrate the correlation between pharmacological effects and drug toxicity. Novel targets for anticancer drugs have been suggested recently, thanks to new finding sin tum or cell and molecular biology such as molecular regulations of cell division, apoptosis, macromolecular rocessing, in vasion, and angiogenesis. Since many of these molecular targets are ounding oncancerous tissues as well, it is important to ensure that any unintended toxicity in healthy tissue is considerably lower than in the cancerous cells, or to provide a justification for why the target is still relevant even if there maybe some toxicity to normaltissues(i.e.monitoringandmanagingdamagetohealthytissue)1.Aninterestingcase of an unintended pharmacology related adverse effectis associated with the biologic in fliximab—a monoclonal antibody against tumor necrosis factor (TNF), indicated for the treatment of rheumatoid arthritis and Crohn’s disease. The inhibitory effects of infliximab on macrophage activation are the mechanism for its desired anti-inflammatory effects. However, diminished macrophage activities cause an increased susceptibility of the patients towards infection. A warning was added to infliximab in 2001for the following reason as stated in a letter from the manufacturer to healthcare professionals: “The Box Warning was added as a resul to the occurrence of 84 case soft tuberculosis worldwide, during the period from August 24th, 1998, through June30th,2001...An increased risk of infections associated with tumor necrosis factor blockade, is consistent with the known effects of TNF on macrophage activation and granuloma formation1.” One should also anti cipate possible drug–drug interactions based on pharmacological properties. A recent case of pharmacological drug–drug interaction is the interaction between sildenafil, a drug for erectile dysfunction. Sildenafil acts via the inhibition of cGMP specific type 5 phospho di esterase (PDE5). It also produces mild decreases in systolic and diastolic blood pressure and an array of minimal side effects, probably due to the inhibition of other types of phosphor di esterase. Drug interactions involving the concurrent use of sildenafil with nitrates and nitrites can produce profound hypotension leading to decreased coronary perfusion and myocardial infarction. A May 1998 letter from the drug manufacturer 7 warns that the drug is not to be co-administered with organic nitrates. This potentially fatal drug interaction also led to the withdrawal of several sildenafil-containing herbal A conscientious effort to evaluate pharmacologically relatedad verse drug effects should allow one to avoid the selection of aproblematic target and to identify managements trategies early on to eliminate unexpected post marketing adverse events. Examples of pharmacology- related toxicological investigations are as follows9:
Chemistry
Many times during drug discovery, a project is abandoned because the major chemical structure (scaffolding) chosen has undesirable toxicity, which cannot be overcome via modification no the side-chains. It is there for important to make sure that chemical structures with a high probability of success are chosen earlier in the program, especially when multiple chemical structures are found positive in the early screening process for efficacy10. One early approach is to evaluate whether the major chemical structure chosen has a history of safe tyre late d problems. In silico approaches continue to be developed to correlate chemical structure with toxicity. As of this writing, it is generally believed that in silico approaches are adequate for the prediction of genotoxicity such as the Ames Salmonella/ histidine - revision assay, but are not yet applicable for other types of toxicity (e.g. hepato toxicity; cardiotoxicity) 11. A promising approach is to perform in vitro toxicological assays early in drug discovery to allow the selection of the chemical structures with the least toxicological liabilities. Combined use of efficacy screens and in vitro toxicity screens allows one to evaluate whether the chemical structures important for efficacy (pharmacophores) can be distinguished from those responsible to toxicity (toxicophores)12.
Examples of chemistry-related toxicological questions are as follows:
1. Are there known adverse drug effects associated with the major chemical structure (scaffolding)? 2. 2. Are there chemical side-chains with known toxicity (structural alerts for toxicity)?
3.Drugmetabolismand pharmacokinetics
The relationship between drug metabolism and toxicity cannot be overemphasized. Metabolism-related toxicity is responsible for a number of adverse drug effects in the liver, the organ where first past drug metabolism occurs. Species-differences in drug metabolism represent a key reason for species-differences in drug toxicity. Pharmacokinetic drug–drug interaction, the effect of one drug on the metabolic clear anceo faco-administered drug, is Also a major mechanism of adverse drug effects. While or gan specific toxicity can be a function of metabolism in specific organs (e.g. liver, kidney), it also can be due to bioaccumulation (e.g. CNS toxicants)14. An important development in drug metabolism is the general acceptance of human tissue- derived systems, especially human liver-derived systems such as liver microsomes and fresh and cryopreserved human hepatocytes, in the evaluation of human drug metabolism. Such studies include the evaluation of intestinal uptake, metabolic stability, metabolite identification and pharmacokinetic drug–drug interactions15. Drug metabolism data obtained with human in vitro system provides critical safety information such as the identification of toxification and detoxification pathways. Drug metabolism data are routinely used to guide the selection of the most “relevant” animal species for safety and pharmacology studies based on their similarities to human in metabolism. In vitro human systems allow one to develop human metabolism data before a drug candidate is administered to humans in vivo16.
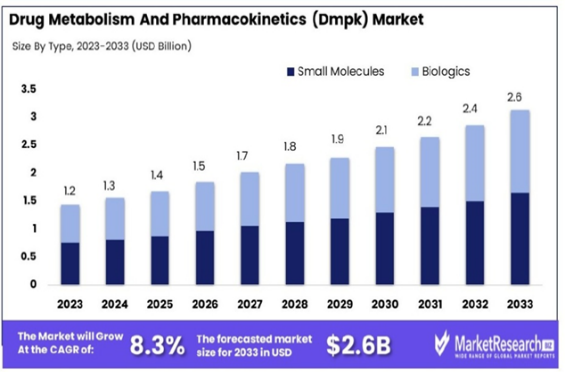
FIG.4.Drug Metabolism And Pharmacokinetics (Dmpk) Market
A consensus is being reached on drug metabolism properties, which appear to occur frequently in drugs with fatal idiosyncratic drug toxicity. These properties include the formation of reactive metabolites, enzyme induction, P450-related toxification pathways and propensity for drug–drug interactions 17. These common properties are consistent with the proposed mechanisms of idiosyncratic drug toxicity and can be used to guide the elimination of drug candidates with high probability of causing idiosyncratic drug toxicity18
Examples of drug metabolism and pharmacokinetics-related toxicological questions are as follows:
4.Toxicology
Well-designed toxicity studies are key to safety assessment. The commonly applied approach of a battery of geno toxicity assays, and studies with laboratory animals including acute, sub chronic and chronic studies, developmental toxicity studies, and life-time carcinogenicity assays are invaluable in the evaluation of drug toxicity. The emphasis here is that the toxicity observed should be evaluated mechanistically to derive the most accurate prediction of human safety19. Although the definition of human safety is the ultimate goal, it is also important, during early phases of drug development, to predict animal toxicity that may occur during preclinical safety trials to allow one to design the most effective animal studies. A key in the use of laboratory animals is to use species that are most “relevant” to human, whenever possible. Key toxicity determinants that are different between the laboratory animals used and humans should be clearly defined to aid data interpretation20 An expert group recently concluded that human based experimental systems are useful in aiding the prediction of human drug toxicity and that in vitro systems with primary cells, especially from human organs, serve as promising experimental systems for the evaluation of human-specific drug properties . Examples of such assays are the use of human blood vessel endothelial cells in the evaluation of vascular toxicity , human hepatocytes in the evaluation of hepato toxicity, and human kidney proximal tubule cells for nephro toxicity. A most recent development is an integrated multiple organ culture system which integrates cells from multiple organs for the evaluation of drug toxicity . An advantage of in vitro experimental systems using primary cells, which retain human specific properties is that the results are likely to be relevant to human. A caveat of the use of in vitro systems is that care must be taken to avoid erroneous conclusions due to in vitro artifacts and the performance of experiments under physiologically irrelevant conditions (e.g. dose levels that would not be achievable in human in vivo), and to fully recognize the limitations of the in vitro system(e.g. the lack of blood circulation, excretion, multiple organ and tissue interactions (which may be improved via the use of the integrated co-culture system, idMOC (, and an intact immune system)21. Toxicology studies should be performed using an investigative approach. Adverse effects should be further defined mechanistically, using endpoints and experimental systems, which may not be routine. In vitro approaches using primary cells from human or animal organs, high content assays such as genomics, proteomics, metabolomics, are examples of experimental tools for mechanistic studies. An example of an application of novel technologies is a recent study with troglita zone, a drug successfully marketed for the treatment of type II diabetes but was withdrawn due to its association with fatal liver toxicity. Troglitazone was found to induce a significantly higher number of gene expression changes forabatteryoftoxicologicallyrelevantgenesthantherelativelynontoxicstructureanalogs rosiglitazone and pioglitazone . Based on the differences between toxic and nontoxic compounds in their effects on gene expression, one can construct possible mechanisms of toxicity, which can be experimentally verified and applied towards the prediction of human effects. Additionally, the knowledge can be applied for the development of biomarkers of toxicity and the development of screening assays for specific toxic liability22.
Examples of toxicological questions that are relevant to the prediction of human drug toxicity are listed below:
While it is true that dose is key to toxicity, the dose that is toxic to different individuals may differ due to physiological, environmental and genetic factors. A dose that is nontoxic to a majority of the patient population may be fatal to an individual due to one or more of these factors (risk factors).A casein point, the analge sicacet amino drug but is known to cause fatal hepatotoxicity, especially in individuals who consumed alcohol. Alcohol has been identified as a risk factor for acetaminophen, presumably due to the induction of the metabolic “toxification” pathway (e.g. cytochrome P450 is form 2E1) as well as the reduction of detoxifying protective cofactors (e.g. reduced glutathione)5.
The risk factor approach in drug toxicity is implied in a recent proposed hypothesis for idiosyncratic drug toxicity, the Multiple Parameter Hypothesis, which states that the low frequency of idiosyncraticdrugtoxicityisduetocon Currence of multiple in dependent events. Based on the hypothesis, the probability for idiosyncratic drug toxicity(Pidt) is a product of the following independent probabilities: (1)exposure to the drug (e.g. dose; Pexp); (2) inherent biological properties of the drug due to its chemical structure (e.g. ability to form reactive metabolites; Pchem); (3) environmental risk factors (e.g. co-exposure to interacting foods or drugs ;Penvir on);and (4)host risk factors(e.g. genetic determinant for drug toxicity; disease conditions predisposing an individual to drug toxicity; Phost):
Pidt= Pexp Pchem Penviron Phost
A corollary of the Multiple Parameter Hypothesis is that there exist risk factors that can dramatically enhance a drug’s toxic potential. Individuals in an environment at a specific point in time may have the “right” combination of risk factors that, if administered a drug with idiosyncratic toxic properties, would succumb to its toxicity11. where Tox individual is the toxicity of the drug in a particular patient at the specific time of administration (e.g. dose to cause liver failure); D the dose of the drug administered; Tox inherent the inherent toxicity of the drug as related to the chemical structure; and RF total represents a single risk factor as a result of all risk factors which can be physiological, environmental and genetic factors that the patient has or is subjected to that would enhance toxicity15. Definition of risk factors should be based on the mechanistic understanding of the key toxic pathways. For instance, if toxicity is due to the formation of toxic metabolites, one needs to define potential risks due to individual with enhanced toxification and/or reduced detoxification pathways as well as pharmacokinetic drug–drug interactions that may increase a patient’s body burden of toxic metabolites As of this writing, risk factors associated with drug metabolism (e.g. induction of toxifying metabolic activities; reduction of detoxifying activities; polymorphism of metabolic enzymes; pharmacokinetic drugdrug interactions) probably are better defined than risk factors not associated with drug metabolism . Current investigation of nonmetabolic risk factors for established drugs (e.g. inflammation, disease status) should help define risk factors of new drugs. There is evidence, for instance, that inflammation is a risk factor for drug induced liver failures 8.
A thorough understanding of risk factors based on known pharmacology, chemistry, drug metabolism, toxic mechanism, and patient characteristics will aid key decisions in drug development. An estimation of the probability of human populations with unfavorable risk factors (to allow a go/no-go decision), and the feasibility of the identification of at-risk populations (to allow safe administration of the drug), are information, which may be critical to the development of safe drugs. Examples of questions regarding risk factors are list edhere:
B. Implementation
The proposed comprehensive approach allows one to assess drug safety intelligently and scientifically, and therefore should be an integral part of the drug discovery and development process, from target selection to clinical trials. Drug candidates conscientiously selected based on the implementation of this approach should have a higher probability of clinical success than drugs selected based mainly on efficacy alone25 This comprehensive approach is best practiced by a team with members with in depth knowledge of the multiple scientific disciplines described (pharmacology, chemistry, drug metabolism, pharmacokinetics, toxicology, genetics). An example of a Comprehensive Drug Safety Evaluation Team is one led by a toxicologist, with team members with expertise in pharmacology, chemistry, drug metabolism/pharmacokinetics, pathology, genetics and supplemented by other scientific disciplines (e.g. epidemiology, statistics, medicine) as needed. Two major objections to the adoption of this comprehensive approach to drug safety evaluation are as follows:
The underlying principle for the proposed approach is that the accuracy of drug safety can be enhanced via a multidisciplinary collaboration to allow a clear understanding of toxicity- related drug properties including pharmacology, chemistry, drug metabolism, toxicology and risk factors. Recent advances in informatics, high content assays such as genomics, proteomics, metabolomics, in vitro biochemical, molecular and cellular experimental systems, should aid this comprehensive approach to evaluate drug safety As drug toxicity is a key determinant of success, secondary only to efficacy, it should be an integral part of drug discovery and development. It is envisioned that the proposed integrated, multidisciplinary approach will enhance the efficiency of drug development via minimizing the probability of the development of drugs with unacceptable toxicity. Most of the studies outlined in this proposal are already being executed in most drug development programs—this approach simply place human drug toxicity as the major focus and driving orce. Adaptation of this approach will no doubt involve more resources than the current “routine” approach, but it is expected that the end should justify the means—the minimization of costly outcomes such as clinical failures and market withdrawal due to adverse drug properties should more than compensate for the extra initial expense16.
Finally, the following modified version of the Paracelsus’ Principle is proposed:
“While dose makes the poison, environmental, genetic, and physiological factors determines the dose that makes the poison for an individual”
The environmental factors include co-administered foods, drugs and environmental chemicals; genetic factors include drug metabolizing enzyme genes and various damage– repair genes; and physiological factors include size, age, gender and disease states26. Accurate prediction of human drug toxicity requires not just the analysis of dose–response relationship, but also a clear knowledge of the mechanism of toxicity and the corresponding risk factors15.
CONCLUSION
While much progress has been made in PV practices, many deficiencies and issues still exist in the efforts to ensure safe medicine usage. Harmonization of PV practices be yon dregulation requires defining and implementing “best suitable practices” for the health-care professionals, industry and the regulatory authorities. It requires formal training for PV professionals and better communication tools. Safety information is communicated between different regulatory agencies, regulatory agencies and manufacturers, health care professionals and manufacturers, agencies and healthcare professionals, healthcare professionals and consumers. All parties in communication utilize different tools– from product labeling to adverse event reports. In today's technological environment these communications are occurring more frequently over the internet, through social media and the cloud. For PV practices to become truly global, there is a further need to integrate these PV best practices with these new modes of communication. Identifying the discrepancies in existing practices is also only a first step. More work is required to establish the best practices, tools and infrastructure that will be required to address the needs of PV in the future. International organizations must continue to advance their understanding of PV and establish guidelines for shifting away from a focus on finding harm and more toward extending knowledge about safety to all appropriate stakeholders. Wallace and Evans write, “Pharmacovigilance should operate in a culture of scientific development. This requires the right balance of inputs from various disciplines, a stronger academic base, and greater availability of basic training and resource which is dedicated to scientific strategy.” Of course, implementing such strategies will require legislative change; thus, the process that begins with the legislation to identify where disharmony exists, must also end with the legislation to create a framework at a national level that allows for an international harmonization of practice.
REFERENCES


Sainath Marakwad*, Tejeshwini Dhawle, Dr. Gajanan Sanap, A Comprehensive Approach for Drug Safety Assessment, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 1, 648-658. https://doi.org/10.5281/zenodo.14627600
 10.5281/zenodo.14627600
10.5281/zenodo.14627600
