Department of Pharmaceutical Chemistry, College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram-695011, Kerala, India.
Thiazolidinediones (TZDs) are a well-known class of antidiabetic agents predominantly recognized for their action as peroxisome proliferator-activated receptor gamma (PPAR-?) modulators. Despite their role in improving insulin sensitivity, the clinical use of TZDs is limited by notable side effects like fluid retention, weight gain, liver toxicity, and an increased risk of heart failure. However, emerging evidence suggests their potential to affect multiple biochemical targets beyond PPAR-? activation. This review focuses on the various antidiabetic mechanisms of TZDs, encompassing their involvement in inhibiting key targets such as ?-glucosidase, ?-amylase, aldose reductase, Protein tyrosine phosphatase 1 B (PTP1B), and Dipeptidyl peptidase-4 (DPP-4), as well as their emerging role as mitochondrial uncouplers. Understanding the mechanisms helps in designing newer agents that could improve the safety as well as ameliorate the off-target effects of existing thiazolidinediones. This review also offers a foundation for future drug development and therapeutic opportunities of TZDs in managing complex metabolic disorders like diabetes.
Diabetes mellitus (DM) is a prevalent metabolic condition marked by consistently high levels of blood sugar [1]. The two primary causes of hyperglycaemia are inadequate insulin secretion and/or insulin resistance [2,3]. As per the International Diabetes Federation, in 2022, approximately 464 million individuals globally were affected by diabetes, with type 2 diabetes accounting for nearly 90% of all cases [4]. The key strategies involved in type 2 DM management are blood glucose regulation and insulin sensitization [5]. The rising prevalence of type 2 DM presents a major health challenge, leading to complications such as retinopathy, nephropathy, coronary artery disease, etc., necessitating the urge to introduce medications like α-glucosidase inhibitors, glinides, biguanides, and glitazones [6,7]. Since single-drug regimens are often insufficient for managing complex metabolic diseases like diabetes, a multitargeted therapeutic approach is preferred [8]. Thiazolidinediones (TZDs), also known as glitazones, are a well-known class of antidiabetic medications that act as a positive modulator of PPAR-γ receptors [9]. These are a class of heterocyclic compounds with a 5-membered saturated ring, containing sulphur and nitrogen atoms at the 1st and 3rd positions respectively. In addition to this, there are two carbonyl groups at the 2nd and 4th positions [10-13]. TZD scaffold can be substituted at the 3rd and 5th positions by different moieties to make it more potentially active [14]. 2,4-TZD core is often combined with diverse heterocyclic substitutes, resulting in the generation of distinct lead compounds which are investigated for their potential to act as antidiabetic, anticancer, antimicrobial, anti-inflammatory, antimalarial, and antioxidant agents [15-19]. Recent studies have demonstrated that TZDs exert their antidiabetic effects through multiple targets. This review focuses on the different antidiabetic targets of TZDs beyond their action as a PPAR-γ modulator.
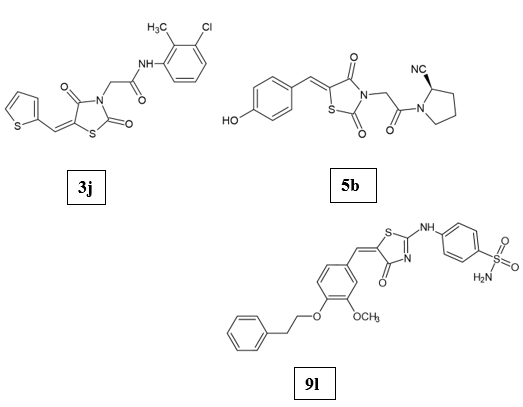
Peroxisome proliferator-activated receptors (PPARs) are a group of transcription factors activated by ligands and belong to the nuclear receptor family. They control the expression of specific target genes by forming heterodimers with retinoid X receptor (RXR) and attaching to particular PPAR response elements (PPREs) found in the promoter regions of those genes [20-22]. When activated by agonists, PPAR-γ improves insulin sensitivity and promotes glucose uptake in individuals with diabetes [23]. Mahendra Gowdru Srinivasa et.al., developed a series of novel 2,4-thiazolidinedione derivatives (3h-3j) as PPAR-γ modulators for antidiabetic activity. The structure of the synthesized compounds was confirmed via spectroscopic techniques. Molecular docking and dynamics simulations were used to investigate the binding modes and stability of the compounds with the target protein. The compound 3j with 3-Chloro-2-methylphenyl substitution had the highest binding score of -7.765 kcal mol-1. ADMET analysis showed that the compounds satisfy Lipinski’s criteria for drug likeness. The compounds were determined to be non-carcinogenic. Cytotoxicity assays performed on C2C12 myoblast cells indicated non-toxicity of the compounds at concentrations of 100 μM and 200 μM. The results of the glucose uptake assay demonstrated that the compounds significantly enhanced glucose uptake compared to insulin and pioglitazone. PPAR-γ transcription factor assay showed a dose-dependent increase in the expression of PPAR-γ in all samples. In vivo studies using a dexamethasone-induced diabetic rat model demonstrated the blood glucose-lowering potential of the compounds. Histopathological examination revealed that treatment with the compounds effectively restored tissue changes in diabetic rats [24]. Shriram D. Ranade et.al., employed an integrated approach combining in silico, in vitro, and in vivo methods to develop and assess a series of acetyl pyrrolidine-2-carbonitrile-based substituted benzylidenethiazolidine-2,4-dione hybrids (5a-5j) targeting PPAR-γ receptors. Pharmacophore modeling and screening were followed by the synthesis of the top ten hits using Knoevenagel condensation and further derivatisation. In vitro glucose uptake capacity was evaluated in L6 myotube cell lines and demonstrated the uptake values of 79.29 ± 1.02 % and 74.58 ± 0.54 % for compounds 5b with 4-hydroxybenzaldehyde and 5f with 4-fluorobenzaldehyde substitution, respectively, compared to pioglitazone (82.36 ± 0.98). The compounds showed a dose-dependent increase in PPAR-γ transactivation, followed by amelioration of glucose homeostasis as well as reversal of insulin resistance in fructose-induced rats. Compound 5b had a better half-life of 4.21 h and an elimination constant of 0.381 when compared to 5f [25]. Islam H Ali et.al., reported two novel series (9a-l and 10a-f) of derivatives by combining the thiazoline-4-one and benzenesulphonamide scaffolds as PPAR-γ modulators. The compounds 9b, 9i, 9k, 9l, and 10d exhibited a high percentage of PPAR-γ activation, and these were further tested for their EC50 determination on PPAR isoforms. The compound 9l (phenethyl thiazoline-4-one sulphonamide) showed the highest PPAR-γ activation (41.7 %). The selectivity of the five derivatives was at their highest level to PPAR-γ > PPAR-δ > PPAR-α with EC50 values of 2.94-11.13 μM. Two derivatives, 9i and 10d, displayed no evidence of toxicity, and both compounds were found to improve insulin secretion. The compound 9i showed a better antidiabetic activity than pioglitazone [26].
TZDs as α-glucosidase and α-amylase inhibitors
One of the most commonly recommended treatments for diabetes involves inhibiting the two digestive enzymes responsible for carbohydrate metabolism: α-amylase and α-glucosidase. Both enzymes play a key role in the digestion of complex polysaccharides. α-Amylase breaks down these polysaccharides into oligosaccharides and disaccharides, which are then further hydrolyzed by α-glucosidase in the intestinal lumen before absorption [27]. Therefore, inhibiting these enzymes is crucial in managing postprandial hyperglycemia [28-30]. Chunmei Hu et.al., developed a series (IT1-26) of twenty-six indole-containing thiazolidine-2,4-diones as α-glucosidase inhibitors. The strongest inhibitory activity was exhibited by the compound IT4 with 4-methyl substitution (IC50 value of 2.35 ± 0.11 μM) compared to the standard drug Acarbose (IC50 value of 575.02 ± 10.11 μM). IT4 presented a reversible mixed-type inhibition on α-glucosidase. The interaction between IT4 and α-glucosidase led to static quenching. In vivo studies demonstrated that administration of IT4 could suppress fasting blood glucose levels [31]. Mahendra Gowdru Srinivasa et.al., clubbed thiazolidinedione nucleus with 1,3,4-oxadiazole using a methylene linker to design a series of novel 3-((5-phenyl-1,3,4-oxadiazole-2-yl)methyl)thiazolidine-2,4-dione (5a-5j) and evaluated their potential against α-amylase and α-glucosidase. The most active compound was 5j with 2,4-dihydroxy substitution (binding score of -6.56), which appears to form hydrogen bonds with amino acids Thr1369 and Thr1586 residues. The Structure Activity Relationship (SAR) of the compounds was proposed. The introduction of a methylene linker between the two heterocyclic nuclei enhanced the activity of the compound. The presence of electron-withdrawing groups at ortho and para positions was found to increase the activity, whereas electron-donating groups at the para position decreased the activity of the compounds towards the enzymes. ADME properties were calculated using QikProp and were found to meet the criteria for drug-likeness. On the basis of Osiris property explorer predictions, the compounds showed green colour, which demonstrated that they are safe and there were no signs of toxicity. The compounds 5a (2,4-dichloro), 5b (4-bromo), and 5j (2,4-dihydroxy) were found to be the most potent derivatives with IC50 values of 17.58 ± 0.19 μM, 22.25 ± 0.22 μM, and 17.21 ± 0.22 μM, respectively. Acarbose was taken as the standard drug (IC50 value of 23.73 ± 1.22 μM) [32]. Gurpreet Singh et.al., synthesized and evaluated nineteen (7a-7s) 3,5-disubstituted derivatives of thiazolidinediones as dual inhibitors of α-amylase and α-glucosidase. Compound 7p with 4-hydroxy and 3-ethoxy substitution emerged as the most potent inhibitor with an IC50 value of 10.33 ± 0.11 μM for α-glucosidase and 10.19 ± 0.25 μM for α-amylase. These results demonstrated that 7p was more active against α-amylase when compared to α-glucosidase. It also displayed notable antioxidant properties. Docking studies revealed strong binding of the compounds within the active site of the target protein. In vivo studies conducted in Wistar rats (STZ-induced diabetic model) showed that 7p decreased blood glucose levels. Therefore, it could emerge as a potential drug candidate against diabetes [33].

TZDs as aldose reductase (ALR2) inhibitors
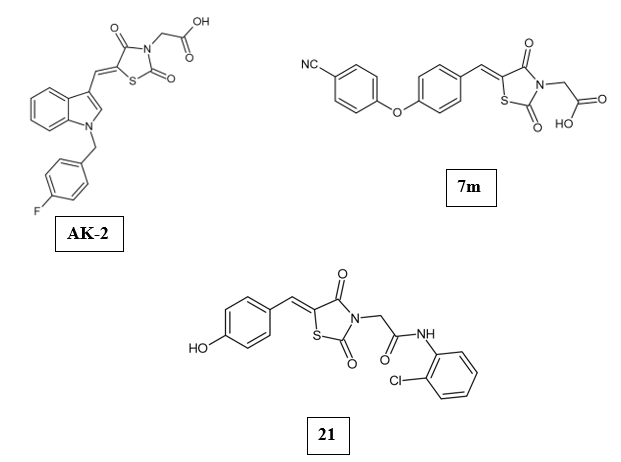
Aldose reductase (ALR2) is the key enzyme of the polyol pathway involved in the pathology of chronic diabetes-related complications. Several aldose reductase inhibitors have demonstrated their ability to curb the accumulation of sorbitol and thereby slow the progression of diabetic complications. However, the development of drugs with sufficiently strong inhibitory activity has been hindered by undesirable pharmacokinetic properties and unintended interactions with off-target molecules that induce toxic side effects. Continued research aims to design novel aldose reductase inhibitors that can avoid these pitfalls and realize the clinical potential of this therapeutic approach for managing the consequences of diabetes [1]. Antonios Kousaxidis et.al., rationally designed and analyzed novel N-benzylindole-based epalrestat analogs (AK1-4) against the aldose reductase enzyme. An indole ring was introduced into the structure of epalrestat (a known aldose reductase inhibitor), ensuring that the 5-ene substitution on the thiazolidinedione maintains a Z-configuration, to replicate its binding interaction within the enzyme’s active site. Among the synthesized derivatives, AK-2 (4-fluoro substitution) emerged as the most potent inhibitor of ALR2 from rat lenses, with an IC50 value of 18 μΜ and a selectivity factor of 787 against ALR1 from rat kidneys [34]. Nagesh Patnam et.al., developed a series of thiazolidinedione derivatives (6a-o and 7a-o) and tested their effectiveness against aldose reductase. Changing ester derivatives (6a-o) to their acid forms (7a-o) showed an increase in antidiabetic potential. All derivatives have shown a remarkable binding affinity towards the enzyme. Most interestingly, the compound 7h had the highest binding energy of -13.7 kcal/mol. From the two series of compounds, 7m was the best characterized with an IC50 value of 0.12 ± 0.14 μM and was tested to be a far superior inhibitor to the reference drug Sorbinil (IC50 value of 2.01 ± 0.25 μM) [35]. Mohd Usman Mohd Siddique et.al., explored a set of 33 N-substituted benzylidene thiazolidinedione derivatives as possible non-carboxylic inhibitors of aldose reductase with the objective of overcoming the shortcomings of carboxylic acid derivatives like Epalrestat that have suboptimal pharmacokinetic profiles. Compound 21 had the highest potency and selectivity as an ALR2 inhibitor with an IC50 of 0.95 μM for ALR2 and 14.2 selectivity over ALR1. SAR analysis revealed that the presence of a para-hydroxy group on the benzylidene ring and electron-withdrawing ortho-positioned groups on the acetanilide moiety, like chlorine, contributed to enhanced selectivity and activity. Molecular docking and MD simulations showed that compound 21 is a more stable and deeper binder in ALR2’s active site, attributed to greater Van der Waals and π-π stacking interactions. Compound 21 exhibited favorable drug-likeness and was classified as a BCS Class 1V compound (low solubility and low permeability). [36].
2.4 TZDs as PTP1B inhibitors
Protein tyrosine phosphatase 1B (PTP1B) negatively regulates insulin signaling by dephosphorylating tyrosine residues on insulin receptors and their substrates [37,38]. Overexpression of PTP1B curtails the activity of protein tyrosine kinases, impeding insulin receptor activation [39,40]. This disruption in signaling promotes the development of insulin resistance, which is a key factor in the onset of type 2 DM. Thus, PTP1B is regarded as a major therapeutic target for diabetes management [41-45]. Mengyue Li et.al., developed a series (MY1-41) of thiazolidinedione derivatives featuring a 1,3,4-thiadiazole structure and assessed their efficacy against PTP1B. Among these, compound MY17 demonstrated the highest potency (IC50 0.41 ± 0.05 μM) and was identified as a reversible, non-competitive inhibitor of PTP1B. In HepG2 cells, treatment with MY17 was effective in reducing insulin resistance induced by palmitic acid by increasing the levels of phosphorylated insulin receptor substrate and protein kinase B. Animal studies showed that administering MY17 orally led to lower fasting blood glucose levels and enhanced glucose tolerance, along with improvements in lipid profiles in diabetic mice [46]. Antonios Kousaxidis et.al., carried out the rational design and synthesis of novel N-benzyl-indole-based epalrestat analogs (AK1-4) and evaluated them against PTP1B. The compounds AK-1 and AK-4 (2,4-dichloro substitution) were found to be the most promising compounds, possessing a greater percentage of inhibition at 10 μM concentration (IC50 value of 42.8 and 21.3 μM). These derivatives had a selectivity factor of 1.1 and 1.3, respectively. The results of the dilution assay demonstrated that these derivatives behave as irreversible inhibitors of PTP1B [34].

2.5 TZDs as DPP-4 inhibitors
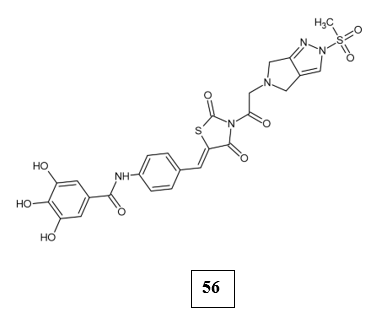
Dipeptidyl peptidase-4 (DPP-4) inhibitors or gliptins work by inhibiting an enzyme responsible for degrading the incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). This inhibition enhances postprandial, glucose-dependent insulin secretion and simultaneously reduces glucagon production [47-49]. Muhammad Shah et.al., investigated the potential of vanillin-morpholine thiazolidinedione hybrid against the DPP-4 enzyme. Structure-based drug design and docking studies were employed to synthesize several new derivatives with modifications on the east (cyclic amines) and west (aromatic substituents) sides of the TZD scaffold. The compound 56, which incorporated a fragment from omarigliptin and gallic acid, was found to be the most potent inhibitor (IC50 value of 0.036 μM). Molecular docking revealed strong binding of the compound within the active site of DPP-4, notably via hydrogen bonding. Glucose uptake and in vivo studies verified the efficacy and safety of the compound, emphasizing its therapeutic value as a novel DPP-4 inhibitor [50].
Mitochondria represent a promising strategy for concurrently managing diabetes and obesity. The process of mitochondrial uncoupling causes energy to be used less efficiently and thereby enhances overall energy expenditure. Thus, uncoupling agents are expected to improve glucose uptake and decrease the amount of fat stored within cells [51]. Antonios Kousaxidis et.al., developed novel N-benzylindole-based epalrestat analogs (AK-1-4), resulting in an unexpected discovery of AK-4 as a mitochondrial uncoupler. AK-4 did not activate the insulin signaling pathway, as confirmed by the Akt phosphorylation status assay. Further analysis revealed that it disrupts the mitochondrial membrane potential, causing an uncoupling effect without reducing mitochondrial mass. ADMET predictions indicated the drug-likeness of the compound, thus representing a promising agent for the treatment of diabetes [34].
Future Prospects
Future research should aim at developing novel TZD analogs or hybrid molecules that retain the beneficial effects while minimizing the off-target issues. Structural optimization to enhance target selectivity, reduce unwanted effects, and foster partial agonism may significantly enhance their safety profile. Such advancements could pave the way for new generation TZD-based agents with improved clinical outcomes and reduced toxicity.
CONCLUSION
Thiazolidinediones (TZDs) have established themselves as effective insulin sensitizers primarily through activation of PPAR-γ. However, recent research revealed that their antidiabetic effects extend beyond this classical mechanism. TZDs also exhibit activity against other critical targets like α-glucosidase, aldose reductase, PTP1B, etc. These multifactorial actions contribute to improved glycemic control and potential benefits in managing diabetes-related complications. Nevertheless, their clinical application is often constrained by adverse effects. While TZDs remain valuable in the pharmacotherapy of type 2 diabetes, their limitations underscore the need for refinement and innovation.
REFERENCES


Revathi K. V.*, Arul K., Thiazolidinediones Beyond PPAR-?: A Review of Their Multifaceted Antidiabetic Actions, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 1196-1207. https://doi.org/10.5281/zenodo.16810059
 10.5281/zenodo.16810059
10.5281/zenodo.16810059
