1Faculty of pharmaceutical sciences, SAGE University Bhopal
2Assistant Professor, SIRT Pharmacy, SAGE university Bhopal, 462043
Computer-Aided Drug Design (CADD) has emerged as a transformative approach in pharmaceutical research, integrating computational tools and molecular modeling techniques to accelerate and optimize the drug discovery process. This review provides a comprehensive overview of the fundamental principles, methodologies, and applications of CADD, including structure-based and ligand-based drug design, molecular docking, pharmacophore modeling, and virtual screening. Advances in bioinformatics, artificial intelligence, and high- performance computing have significantly enhanced the accuracy and efficiency of CADD, enabling the identification of novel therapeutic candidates with improved specificity and reduced development costs. Key case studies illustrating successful drug discoveries aided by CADD are discussed, highlighting its growing impact in both academia and industry. The review also addresses current challenges, such as the limitations of predictive models and the need for better integration with experimental data, while exploring future directions for innovation in this rapidly evolving field.
COMPUTER AIDED DRUG DESIGN
Computer-Aided Drug Design (CADD) is a technology-driven approach that uses computational methods to discover, design, and optimize potential drug candidates by simulating their interactions with biological targets. It plays a crucial role in modern pharmaceutical research by reducing the time and cost associated with traditional drug discovery. CADD includes techniques such as structure-based drug design, which relies on the 3D structure of target proteins, and ligand-based drug design, which uses information from known active compounds. These methods help identify promising molecules with high specificity and efficacy before laboratory testing, making the drug development process more efficient and targeted.
IMPORTANCE OF CADD IN MODERN DRUG DISCOVERY
Computer-Aided Drug Design (CADD) is critically important in modern drug discovery as it significantly accelerates the identification and development of new therapeutic agents. By using computational models to predict how drug candidates will interact with biological targets, CADD reduces the need for costly and time-consuming laboratory experiments. It enables researchers to screen vast libraries of compounds quickly, optimize lead molecules for better efficacy and safety, and identify potential side effects early in the process. CADD also supports personalized medicine by allowing drug design tailored to individual genetic profiles. Overall, it enhances the efficiency, accuracy, and success rate of drug development in both academia and the pharmaceutical industry.
OBJECTIVES OF THE REVIEW
OVERVIEW OF DRUG DISCOVERY PROCESS
1. TRADITIONAL VS. COMPUTER- AIDED APPROACHES
Traditional drug discovery relies heavily on trial-and-error methods, involving extensive laboratory experiments to identify and test new drug compounds. This process is time- consuming, costly, and resource-intensive, often taking over a decade and billions of dollars to bring a drug to market. It typically involves random screening of large compound libraries and iterative chemical synthesis, with limited predictive insight into molecular interactions.
In contrast, Computer-Aided Drug Design (CADD) offers a more targeted and efficient approach by using computational tools to simulate and predict how drug candidates will interact with biological targets. CADD allows virtual screening of millions of compounds in a fraction of the time, guides lead optimization through structure-activity relationship analysis, and reduces the need for excessive experimental work. This not only accelerates the drug development timeline but also improves the success rate by focusing on the most promising candidates early in the process.
2. PHASES: TARGET IDENTIFICATION, VALIDATION, LEAD DISCOVERY, LEAD OPTIMIZATION
Target Identification
This phase involves identifying a biological molecule (typically a protein or gene) associated with a disease. CADD helps by analyzing biological databases and using bioinformatics tools to predict potential targets based on disease mechanisms.
Target Validation
Once a target is identified, its role in the disease is confirmed. Computational modeling and simulations are used to predict the target’s druggability, helping researchers decide if it is suitable for therapeutic intervention.
Lead Discovery
In this stage, compounds that interact with the target are identified. CADD techniques such as virtual screening, molecular docking, and pharmacophore modeling enable rapid identification of potential lead molecules from large chemical libraries.
Lead Optimization
The most promising leads are chemically modified to enhance their potency, selectivity, and pharmacokinetic properties. CADD supports this phase through structure-activity relationship (SAR) analysis, QSAR modeling, and molecular dynamics simulations to predict how modifications affect drug behavior.
TYPES OF CADD APPROACHES
1. STRUCTURE BASED DRUG DESIGN
Structure-Based Drug Design (SBDD) is a method in drug discovery that uses the 3D structure of a biological target, usually a protein, to design molecules that can bind specifically to it. By analyzing the shape and chemical environment of the target's active site, researchers use computer simulations to design or optimize drug candidates for better binding and activity. This approach helps in identifying effective drugs more quickly and accurately, reducing the need for extensive lab testing.
2. LIGAND- BASED DRUG DESIGN
Ligand-Based Drug Design (LBDD) is a computational approach that relies on information from known active molecules (ligands) rather than the 3D structure of the target protein. It is used when the structure of the biological target is unknown or unavailable. By analyzing the chemical and biological properties of active compounds, such as their shape, electronic features, and binding activity, researchers develop models (like QSAR and pharmacophore models) to predict new molecules with similar or improved effects. LBDD is valuable for identifying and optimizing drug candidates when only ligand data is available.
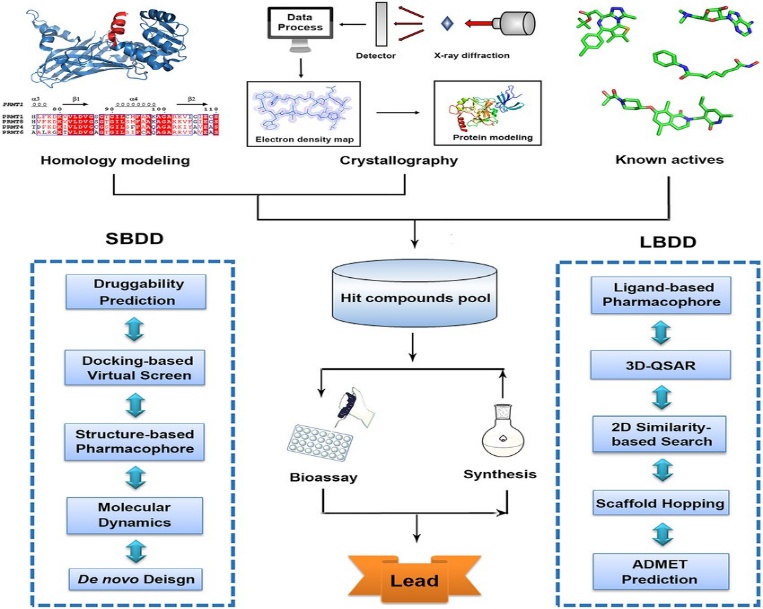
COMPUTATIONAL TECHNIQUES IN CADD
1. MOLECULAR DOCKING AND SCORING FUNCTIONS
Molecular docking is a computer-simulated process used in drug design to predict how a small molecule (ligand) interacts with a target protein at the atomic level. The objective is to find the most favorable orientation and conformation of the ligand when bound to the active site of the target. This helps in understanding binding interactions and identifying potential drug candidates.
Docking involves two key components:
1. Search Algorithm: It explores various possible orientations and conformations (poses) of the ligand within the binding site of the protein.
2. Scoring Function: It evaluates each pose and estimates the binding affinity based on physical and chemical properties, helping to rank compounds by their predicted effectiveness.
Scoring functions can be categorized into:
1. Force-field-based, which use molecular mechanics to calculate interaction energies;
2. Empirical, which are based on regression models using experimental data;
3. Knowledge-based, which use statistical potentials derived from known protein-ligand complexes;
4. Machine learning-based, which predict binding affinities from large datasets using trained algorithms.
2. MOLECULAR DYNAMICS SIMULATIONS
Molecular Dynamics (MD) simulations are computer-based methods used to model and analyze the physical movements of atoms and molecules over time. The technique is grounded in classical mechanics, where Newton’s laws of motion are applied to predict how each atom in a molecular system behaves under the influence of forces from neighboring atoms. In drug design, MD simulations are used to study the dynamic behavior of biomolecular systems such as protein-ligand complexes. Unlike molecular docking, which provides a static snapshot, MD allows researchers to observe how the protein and ligand interact and change shape in a simulated biological environment over time (typically nanoseconds to microseconds). These simulations help assess the stability of the drug-target binding, reveal conformational changes, and identify key interactions important for binding affinity.
3. PHARMACOPHORE MODELING
Pharmacophore modeling is a ligand-based drug design technique that identifies the essential structural features responsible for a molecule’s biological activity. A pharmacophore is defined as the spatial arrangement of features such as hydrogen bond donors or acceptors, hydrophobic regions, aromatic rings, and charged groups that are necessary for optimal interaction with a specific biological target. The goal of pharmacophore modeling is to create an abstract representation of these features based on one or more known active compounds. This model can then be used to screen chemical databases to identify new compounds that fit the pharmacophore and may exhibit similar biological activity, even if they have different overall structures.
4. MACHINE LEARNING AND AI IN CADD
Machine Learning (ML) and Artificial Intelligence (AI) play a crucial role in Computer-Aided Drug Design (CADD) by analyzing large datasets to predict drug-target interactions, screen compounds, optimize lead molecules, and forecast ADMET properties. These technologies enhance the speed, accuracy, and efficiency of drug discovery by identifying patterns and making data-driven predictions that traditional methods might miss. AI models are especially useful in tasks like virtual screening, QSAR modeling, and de novo drug design, significantly accelerating the development of new therapeutics.
DATABASES AND RESOURCES
1. PROTEIN DATA BANK, DRUGBANK, PubChem, ChEMBL
• Protein Data Bank (PDB):
A comprehensive repository of 3D structural data for biological macromolecules such as proteins and nucleic acids. It is widely used in structure-based drug design for analyzing protein-ligand interactions and preparing targets for molecular docking.
???? Website: https://www.rcsb.org
• DrugBank:
A database that combines detailed drug data with comprehensive information on drug targets, mechanisms, interactions, and chemical properties. It is valuable for drug repurposing, target identification, and pharmacological profiling.
???? Website: https://go.drugbank.com
• PubChem:
A public chemical database maintained by the NCBI, containing information on millions of compounds, their structures, biological activities, and properties. It is used for virtual screening, compound sourcing, and structure-activity analysis.
???? Website: https://pubchem.ncbi.nlm.nih.gov
• ChEMBL:
A large bioactivity database maintained by EMBL-EBI, containing information on drug-like molecules and their bioactivities against various biological targets. It supports QSAR modeling, lead identification, and SAR studies.
???? Website: https://www.ebi.ac.uk/chembl
2. MOLECULAR LIBRARIES AND CHEMICAL DATABASES
Molecular libraries and chemical databases are fundamental components of Computer-Aided Drug Design (CADD). They consist of large collections of chemical compounds, either real or virtual, along with associated structural, physicochemical, and biological data. These resources are used to identify potential drug candidates through techniques such as virtual screening, pharmacophore modeling, and QSAR analysis. Chemical databases typically include information such as molecular structures, SMILES strings, molecular weights, solubility, and bioactivity data against various biological targets. Molecular libraries may also contain compounds that are synthetically accessible or commercially available for experimental validation.
CHALLENGES AND LIMITATIONS
1. INACCURACIES IN TARGET STRUCTURE OR DOCKING SCORES
1. Inaccuracies in Target Structure: Docking simulations often rely on protein structures obtained from experimental techniques like X-ray crystallography or cryo-electron microscopy. These structures may contain errors such as missing residues, flexible loops, or incorrect side-chain orientations. Additionally, proteins are dynamic in nature, but most docking methods treat them as rigid, which can overlook important conformational changes that occur during binding. Homology-modeled structures, used when no experimental data is available, may further introduce structural inaccuracies due to low sequence identity with template proteins.
2. Inaccuracies in Docking Scores: Scoring functions are mathematical models used to estimate the strength of ligand binding. These functions often simplify complex interactions and may fail to account for solvation effects, entropic contributions, or protein flexibility. As a result, they can produce false positives (predicting strong binders that are inactive) or false negatives (missing real binders). Moreover, different docking tools use different algorithms and scoring criteria, which can lead to inconsistent or non-reproducible results.
2. NEED FOR EXPERIMENTAL VALIDATION
In Computer-Aided Drug Design (CADD), computational methods such as molecular docking, virtual screening, and pharmacophore modeling are valuable for predicting potential drug candidates. However, these predictions are based on theoretical models and assumptions that may not fully capture the complexity of biological systems. Therefore, experimental validation is essential to confirm the accuracy and reliability of these computational results.
FUTURE PROSPECTS
The future of Computer-Aided Drug Design (CADD) is promising, with ongoing advancements in computational power, artificial intelligence, and structural biology expected to further revolutionize drug discovery. Integration of machine learning (ML) and deep learning models will enhance the accuracy of predictions in molecular docking, binding affinity estimation, and ADMET profiling. These AI-driven approaches can analyze massive datasets quickly and uncover patterns that traditional methods might miss. The development of quantum computing also holds potential for solving complex molecular simulations with unprecedented speed and precision. Meanwhile, improvements in protein structure prediction (e.g., AlphaFold) are providing high-quality models for previously uncharacterized targets, expanding the scope of structure-based drug design. Additionally, cloud computing, automation, and multi-omics integration will support more collaborative, efficient, and personalized drug discovery efforts. These technologies are expected to reduce the cost and time of drug development, increase success rates, and enable the design of targeted therapies for complex and rare diseases.
CONCLUSION
1. IMPACT OF CADD IN MODERN DRUG DISCOVERY
Computer-Aided Drug Design (CADD) has transformed modern drug discovery by:
1. Speeding up target identification using modeling and simulations.
2. Reducing costs and time via virtual screening and predictive tools.
3. Improving accuracy in lead optimization and binding predictions.
4. Predicting ADMET properties early to reduce clinical trial failures.
5. Enhancing success rates with structure-based and ligand-based design.
6. Supporting AI integration for smarter, faster molecule discovery.
7. Accelerating response to health crises (e.g., COVID-19 drug repurposing).
2. GROWING ROLE AND POTENTIAL FUTURE CONTRIBUTIONS
Growing Role of CADD
Future Potential
REFERENCES


Apurva Patel, Astha Sachdeva, Computer Aided Drug Design, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 2645-2651. https://doi.org/10.5281/zenodo.15432398
 10.5281/zenodo.15432398
10.5281/zenodo.15432398
