P. Wadhwani College of Pharmacy, Yavatmal.
The present study focused on the formulation and evaluation of lamivudine-loaded floating tablets aimed at controlled drug delivery. Physical and analytical characterization confirmed the identity and purity of lamivudine, with the standard calibration curve exhibiting excellent linearity (R² = 0.9992). Solubility and IR studies revealed that lamivudine is soluble in water and chemically compatible with the selected polymers. Among various microsphere formulations (F1–F10), batch F10 showed the highest percentage yield (23%), indicating optimized formulation conditions. Pre- and post-compression evaluations demonstrated acceptable micromeritics and mechanical properties of tablet blends. In-vitro drug release studies revealed controlled release over 12 hours, with batch F1 achieving 99.24% release, governed predominantly by Korsmeyer-Peppas kinetics, suggesting diffusion and erosion mechanisms. Floating lag time was within acceptable limits for all batches. Accelerated stability testing over 30 days confirmed that the physical and chemical properties of selected formulations (F1 and F5) remained stable. Overall, the formulations displayed promising results for controlled oral delivery of lamivudine.
Oral drug delivery remains the most preferred and convenient route of drug administration due to its non-invasiveness, ease of administration, and patient compliance. However, conventional oral dosage forms often face challenges such as rapid gastrointestinal transit and short gastric residence time, which can result in incomplete drug absorption and reduced bioavailability, particularly for drugs that are primarily absorbed in the stomach or upper small intestine. To overcome these limitations, Gastroprotective Drug Delivery Systems (GRDDS) have been developed, which aim to prolong the residence time of dosage forms in the stomach. Among various approaches under GRDDS, Floating Drug Delivery Systems (FDDS) have gained significant attention due to their simplicity and effectiveness in achieving gastric retention. FDDS are designed to have a lower density than gastric fluids, allowing them to float on the stomach contents and remain in the gastric region for an extended period without affecting the normal gastric emptying process. The floating mechanism can be achieved through various designs, such as effervescent systems, non-effervescent systems, and systems incorporating gas-generating agents. These formulations help in maintaining the dosage form in a buoyant state, which ensures prolonged drug release at the desired site of absorption. FDDS are particularly useful for drugs with a narrow absorption window, drugs that are unstable in the intestinal or colonic pH, and drugs that act locally in the stomach, such as antacids, antibiotics, and anti-ulcer agents. Recent advancements in materials science and formulation techniques have enabled the development of various floating systems including floating tablets, capsules, microspheres, beads, and films. These systems not only improve therapeutic efficacy but also minimize side effects and reduce dosing frequency, thereby improving patient adherence. In conclusion, Floating Drug Delivery Systems represent a promising and versatile approach for enhancing the performance of oral drug delivery, particularly for drugs with specific absorption characteristics. Continued research and innovation in this area are expected to lead to more effective and patient-friendly gastroretentive therapies.
AIM: design, development and optimization of floating gastro-retentive drug delivery system.
Objectives:
3 Drug Profile:
3.1 Lamivudine:
Lamivudine is a new generation orally active nucleoside analog launched in the U.S.A. for use in combination with zidovudine (AZT) as a first-line therapy for patients with HIV infection. Lamivudine is rapidly converted to phosphorylated metabolites in the body which act as inhibitors and chain terminators of HIV reverse transcriptase (RT), the enzyme required for the replication of the HIV genome. Lamivudine has similar inhibitory potency to RT as AZT but is 10 times less toxic and is active against AZT-resistant strains of HIV.
3.1.1 Molecular Structure:

3.1.2 Description: Lamivudine is a synthetic nucleoside analogue and is phosphorylated intracellularly to its active 5'-triphosphate metabolite, lamivudine triphosphate (L-TP). This nucleoside analogue is incorporated into viral DNA by HIV reverse transcriptase and HBV polymerase, resulting in DNA chain termination.
3.1.3 IUPAC name: 4-amino-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2-one
3.1.4 Brand name: Epivir (GlaxoSmithKline).
3.1.5 Chemical name:
3.1.6 Physical Properties:
3.1.7 Dose of Drug:
3.1.8 Clinical Pharmacology:
3.1.8.1 Pharmacokinetics:
a) Absorption: Lamivudine is absorbed rapidly, with a maximum lamivudine concentration of 0.5 to 1.5 hours following oral dose administration and absolute bioavailability of approximately 82% in adults and 68% in children. Lamivudine can be taken with or without food. While systemic exposure to lamivudine does not change with food intake, taking lamivudine with food slows the absorption rate by 40%. Lamivudine is manufactured as an oral liquid and tablet form and has equitable bioavailability in adults. The solid tablet is preferred in children because bioavailability is 40% lower in children using the oral liquid form. While both doses of the solid tablet have similar steady-state concentrations and therapeutic effects, the larger dose of 300 mg once daily has shown to have larger trough and maximum serum levels and is less consistent throughout the day than the smaller 150 mg tablet taken twice daily.
b) Distribution: Lamivudine is distributed into the total body fluid / extravascular spaces. The half-life can be as long as 15.5 hours for HIV-infected cells and 19 hours for HBV-infected cells. However, viral replication site reservoirs showed varying pharmacokinetics. Lamivudine has been shown to have a longer half-life and higher IC50 after the first dose in seminal mononuclear cells and the female genital tract. Conversely, the cerebral spinal fluid has shown minimal transmission of lamivudine across the blood-brain barrier, especially in adults. Lamivudine freely traverses the placenta, often presenting with exceedingly high concentrations, and is readily found in breast milk.
c) Metabolism: Lamivudine does not undergo metabolism via the CYP450 pathway and minimally binds to plasma protein. Therefore, CYP450 inducers and inhibitors will not affect its metabolism, nor does the drug have many drug interactions with protein-bound medications. While lamivudine has some drug-drug interactions, like interferon-α blockers, ribavirin, zidovudine, and drugs that inhibit MATE-1, MATE-2K, OCT2 transporters, like trimethoprim and indinavir, which increases the plasma concentration of lamivudine, no interactions have clinical significance. One exception is sorbitol, which demonstrates dose-dependent decreases in the maximum serum concentration of lamivudine.
d) Elimination: Lamivudine is eliminated in the urine and secreted as an active organic cation. The mean elimination half-life was 5 to 7 hours. The average clearance of unaltered lamivudine is 71%, with 5% to 10% excreted as trans-sulfoxide. This clearance indicates the necessity for sufficient kidney function in the patient, and dosing should be adjusted accordingly. Dialysis did not significantly increase the elimination of lamivudine to warrant dose modification. Pregnant women have a 22% increase in lamivudine clearance without leading to sub-exposure.
3.2 Mechanism of Action:
Lamivudine is a dioxynucleoside cytosine analog that inhibits viral DNA synthesis via reverse transcriptase DNA chain termination post phosphorylation. Once inside the cell, lamivudine, 2'-deoxy-3'-thiacytidine, is metabolized to the triphosphate form, lamivudine triphosphate (abbreviated as 3TC-TP or L-TP), and the monophosphate form, lamivudine monophosphate (abbreviated as 3TC-MP or L-MP), during intracellular kinase phosphorylation. Both forms, L-TP and L-MP, inhibit viral DNA synthesis. Lamivudine is advantageous as an antiviral drug because it is primarily not recognized by human polymerase as a substrate. As an L-(-)-nucleoside enantiomer instead of a D-enantiomer, active lamivudine (3TC) is not primarily recognized by human polymerases as a substrate but actively competes with natural cytidine triphosphate to inhibit reverse transcriptase DNA synthesis seen in both HIV-1 and HBV infection. The triphosphate metabolite can also weakly inhibit mammalian DNA polymerase (α and β types) and mitochondrial DNA (mtDNA) polymerase. There are no known pharmacokinetic differences related to gender or race.
3.2.1 Pharmacodynamic:
Lamivudine is a nucleoside reverse transcriptase inhibitor (NRTI) that stops the human immunodeficiency virus type 1 (HIV-1) and hepatitis B virus from making DNA (HBV). Lamivudine can be phosphorylated to produce active metabolites that compete for viral DNA incorporation. Lamivudine metabolites function as a chain terminator of DNA synthesis by competitively inhibiting the activity of the HIV reverse transcriptase enzyme via DNA incorporation. Incorporated nucleoside analogues hinder the development of a 5' to 3' phosphodiester linkage, which is required for DNA chain elongation because they lack a 3'- OH group.
3.2.2 Drug interactions:
There are potentially dangerous interactions with other drugs when used in combination.
Antimicrobials: trimethoprim inhibits the excretion of lamivudine.
Antivirals: avoid concomitant use with foscarnet, emtricitabine, and IV ganciclovir.
Cytotoxic drugs: avoid concomitant use with clarithromycin.
Orlistat: absorption may be reduced by orlistat.
3.2.3 Clinical Use:
Lamivudine is indicated for the treatment of HIV when used in combination with other antiretroviral agents. A lower dose than that used to treat HIV is approved for the treatment of HBV.
4. Excipient Profile:
4.1 Sodium bicarbonate:
4.1.1 Nonproprietary Names:
BP: Sodium Bicarbonate
JP: Sodium Bicarbonate
PhEur: Sodium Hydrogen Carbonate
USP: Sodium Bicarbonate
4.1.2 Synonyms:
Baking soda; E500; Effer-Soda; monosodium carbonate; natrii hydrogenocarbonas; Sal de Vichy; sodium acid carbonate; sodium hydrogen carbonate.
4.1.3 Chemical Name and CAS Registry Number: Carbonic acid monosodium salt [144-55-8]
4.1.4 Empirical Formula and Molecular Weight: NaHCO3 84.01
4.1.5 Structural formula:
4.1.6 Functional Category: Alkalizing agent; therapeutic agent.
4.1.7 Description: Sodium bicarbonate occurs as an odorless, white, crystalline powder with a saline, slightly alkaline taste. The crystal structure is monoclinic prisms. Grades with different particle sizes, from a fine powder to free-flowing uniform granules, are commercially available.
4.1.8 Typical Properties Acidity/alkalinity pH = 8.3 for a freshly prepared 0.1 M aqueous solution at 258C; alkalinity increases on standing, agitation, or heating.
Density (bulk) 0.869 g/cm3
Density (tapped) 1.369 g/cm3
Density(true) 2.173 g/cm3
Freezing point depression 0.3818C (1% w/v solution)
Melting point 2708C (with decomposition)
Moisture content Below 80% relative humidity, the moisture content is less than 1% w/w. Above 85% relative humidity, sodium bicarbonate rapidly absorbs excessive amounts of water and may start to decompose with loss of carbon dioxide. NIR spectra Osmolarity A 1.39% w/v aqueous solution is iso-osmotic with serum. Refractive index nD 20 = 1.3344 (1% w/v aqueous solution) Solubility.
4.6.1 Nonproprietary Names
BP: Xanthan Gum
PhEur: Xanthan Gum
USP-NF: Xanthan Gum
4.6.2 Synonyms: Corn sugar gum; E415; Grindsted; Keldent; Keltrol; polysaccharide B-1459; Rhodicare S; Rhodigel; Vanzan NF; xanthani gummi; Xantural.
4.6.3 Chemical Name: Xanthum Gum
4.6.4 CAS Registry Number: [11138-66-2]
4.6.5 Empirical Formula and Molecular Weight: (C35H49O29) n
4.6.6 Molecular Weight: 106
4.6.7 Structural Formula:
4.6.8 Functional Category: Gelling agent; stabilizing agent; suspending agent; sustained-release agent; viscosity-increasing agent.
4.6.9 Applications in Pharmaceutical Formulation or Technology: Xanthan gum is widely used in oral and topical pharmaceutical formulations, cosmetics, and foods as a suspending and stabilizing agent. It is also used as a thickening and emulsifying agent. It is nontoxic, compatible with most other pharmaceutical ingredients, and has good stability.
4.6.10 Description: Xanthan gum occurs as a cream-or white-colored, odorless, free flowing, fine powder.
4.6.11 Typical Properties:
Acidity/alkalinity: pH=6.0–8.0fora1%w/v aqueous solution.
Freezing point: 08 C for a 1 % w/v aqueous solution.
Heat of combustion: 14.6J/g (3.5cal/g).
Melting point: 270 ?C
Solubility: Practically insoluble in ethanol and ether; soluble in cold or warm water.
4.6.12 Stability and Storage Conditions: Xanthan gum is a stable material. Aqueous solutions are stable over a wide pH range (pH 3-12), although they demonstrate maximum stability at pH 4-10 and temperatures of 10-60 ?C.
HPMC K100M:
4.8.2 HPMC K4M:
4.8.3 Non-proprietary name
USP. Hypromellose
BP. Hypromellose
JP. Hydroxy propyl methyl cellulose
4.8.4 Synonyms: HPMC, Methocel, methyl hydroxypropyl cellulose Methylcellulose propylene Benecel MHPC Hydroxypropyl methylcellulose.
4.8.5 Chemical name: Cellulose- Hydroxypropyl Methyl Ether.
4.8.6 Molecular weight: 10000.0-1500000.0
4.8.7 Structural formula:
4.8.8 Functional category: stabilizing agent, suspending agent, tablet binder, viscosity increasing agent, rate controlling polymer for sustained release, film-former.
4.8.9 Solubility: It is soluble in cool water, form a viscous colloid, insoluble in chloroform, ether and ethanol (95 percent.) soluble in mixtures of ethanol, dichloromethane. Mixing contains of methanol and dichloromethane.
4.8.10 Description: Hypromellose is a tasteless and odourless, white, or creamy white granular powder.
4.8.11 Incompatibility: Incompatible with few oxidizing substances. Since it is nonionic and not complex with metallic salts or ionic organics to form practically insoluble precipitate molecule.
4.8.12 Safety: It is a nontoxic and non-irritant, even excessive oral consumption may cause laxative effect.
4.8.13 Application in pharmaceutical formulations: Hypromellose serves as tablets binder and film former for extended-release tablets. It also acts as thickening agent and suspending for topical formulations and is used in ointments and topical gels as an emulsifier, suspending and stabilizing agent. Moreover, it has applications in capsule manufacturing and as a wetting agent for hard contact lenses and adhesive in plastic bandges.
9. RESULT AND DISCUSSION:
9.1 Preformulation studies:
9.1.1 Physical characteristics of drug:
Table No.9: Physical characteristics of lamivudine.
|
Parameter |
Observation |
|
Colour |
White |
|
Odour |
Odourless |
|
Taste |
Bitter |
|
Melting point |
160-161 ? c |
9.1.2 Solubility studies:
Table No.10: Solubility studies.
|
Sr. No |
Solvent |
Solubility of Lamivudine |
|
1) |
Water |
Soluble |
|
2) |
Ethanol |
Slightly soluble |
|
3) |
Methanol |
Sparingly soluble |
9.2 Analytical characterization of drug sample:
9.2.1 Standard curve of lamivudine:
25 mg of drug lamivudine was dissolved in 0.1N HCL buffer and volume was make upto 25 ml to make stock solution. Then 0.1 ml of stock solution was taken and diluted upto 1000 ug/ml of 0.1N HCL buffer to get concentration 1ug/ml and in similar way dilution were made as 1,2,3,4,5 ug/ml respectively and absorbance measured at 280 nm by uv visible spectrophotometer. The absorbance values were plotted against concentration (ug/ml) to obtain the standard calibration curve. It is given in Table No.11.
Table No.11. Calibration of lamivudine in 0.1 N HCL buffer.
|
Sr. No. |
Volume of stock solution |
Concentration(ug/ml) |
Absorbance |
|
1. |
0.1 ml |
1 |
0.052 |
|
2. |
0.2 ml |
2 |
0.097 |
|
3. |
0.3 ml |
3 |
0.151 |
|
4. |
0.4 ml |
4 |
0.206 |
|
5. |
0.5 ml |
5 |
0.258 |
9.2.2 Calibration curve of lamivudine with 0.1 N HCL buffer:
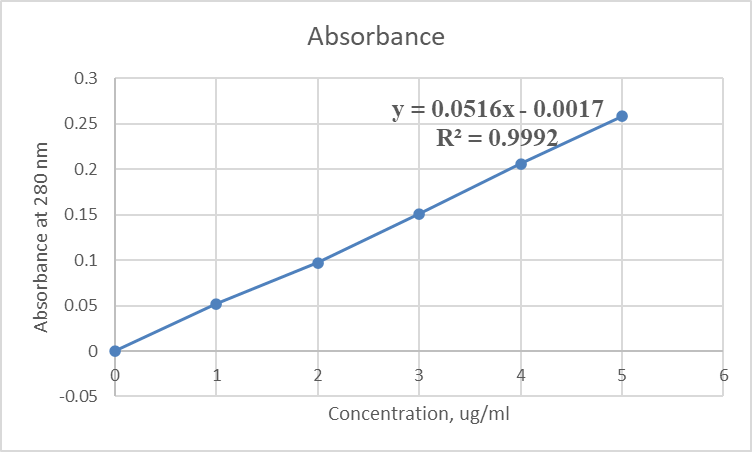
Figure No.12: Calibration curve of lamivudine with 0.1 N HCl buffer.
Correlation coefficient (R2) = 0.9992.
Equation of regressed line = Y = 0.0516x-0.0017.
Where, X = Value of concentration.
Y = Regressed value of Absorbance.
9.3 Evaluation of Spray Dried Microspheres.
9.3.1 Percentage yield (i.e recovery) of Microspheres:
The percentage yield of microspheres was determined by weighing after drying. The bar diagram shows percentage yield of all the formulations F1 to F10 which is corresponds to Table No.12. The percentage yield was found of F10 batch.
Table No.12: Percentage Yield of Lamivudine Microspheres.
|
Batches |
% Yield |
|
F1 |
16.73 |
|
F2 |
18.01 |
|
F3 |
9.44 |
|
F4 |
3.8 |
|
F5 |
7.8 |
|
F6 |
7.39 |
|
F7 |
8.42 |
|
F8 |
9.2 |
|
F9 |
5.28 |
|
F10 |
23 |
% Yield
Lamivudine Microspheres
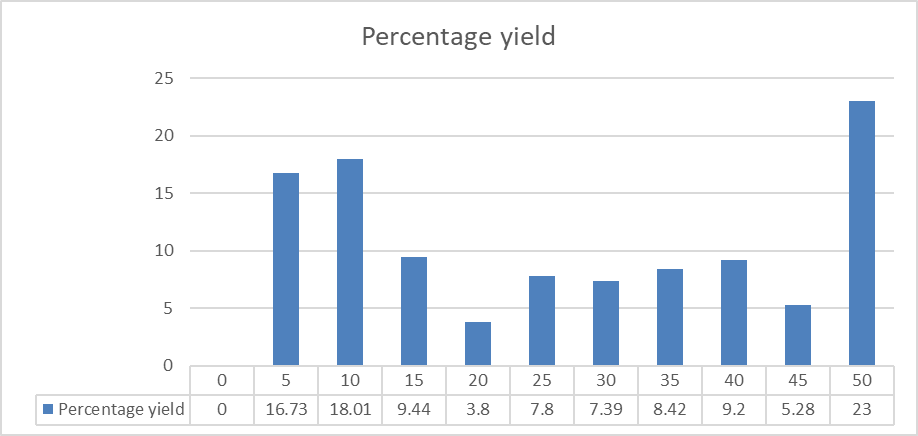
Figure No.13: Percentage yield of formulation F1 to F10.
Observation:
Conclusion:
The study demonstrated that formulation conditions greatly influence the percentage yield of lamivudine microspheres. Batch F10 achieved the highest yield, suggesting optimized formulation parameters in this batch. Further optimization and standardization of the preparation method are necessary to consistently improve the yield across all batches.
9.4 Drug Interaction Studies (Compatibility Studies)
9.4.1 FT-IR Spectroscopy:
Its important to check any kind of interaction between drug and polymers. The polymers which are to be incorporated into formulation should be compatible with the drug. This compatibility study or interaction study was done using Fourier Transformed Infrared Spectroscopy. IR spectra of pure drug lamivudine and polymer Viz. HPMC K 100 M were taken separately. Then to know if there is any interaction between drug and polymer were taken in combination. IR spectra of drug and polymer were taken in combination. The result show that there was seen between drug lamivudine and polymers as there was no significant change in the pattern of peaks of optimized batch.

Figure No.14: IR spectra of pure drug lamivudine.
Table No.13: IR interpretation of lamivudine (pure drug).
|
Sr. No. |
Peaks Observed in IR Spectra of lamivudine |
Functional Group |
|
1 |
3901.99 - 3608.81 |
O-H or N-H stretching (broad peak, H- bonded) |
|
2 |
3331.07-3068.75 |
N-H stretching (amine or amide) |
|
3 |
2837.29 |
C-H stretching (alkane) |
|
4 |
2497.82 |
Possibly S-H stretching (thiol group, weak) |
|
5 |
2281.79-2112.05 |
C≡ 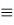
|
|
6 |
1649.14 |
C=O stretching (carbonyl group) or C=N stretching (imine) |
|
7 |
1571.99 - 1510.26 |
C=C stretching (aromatic ring vibrations) |
|
8 |
1402.25 |
C-H bending (alkane or aromatic C-H) |
|
9 |
1340.53 |
C-N stretching (amines) |
|
10 |
1247.94 |
C-O stretching (ether or alcohol) |
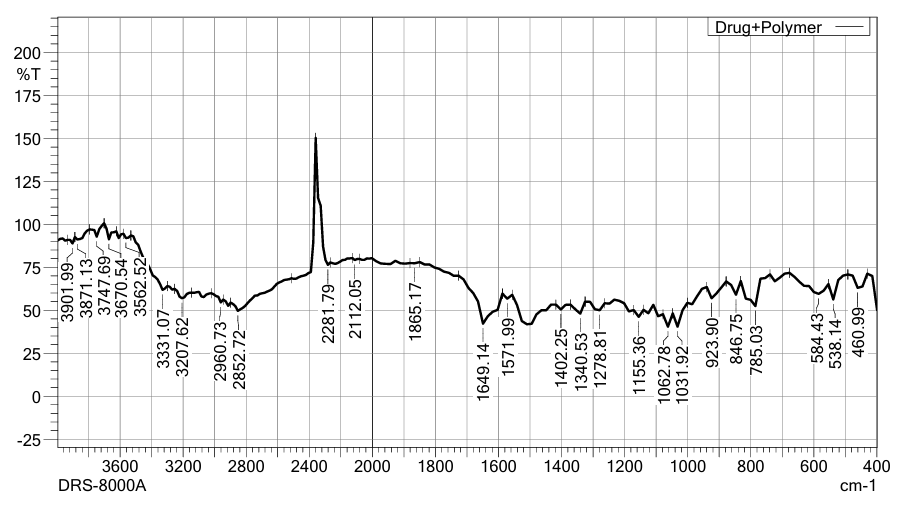
Figure No.15: IR Spectra of Drug + Polymer.
Table No.14: IR Interpretation of drug+ Polymer.
|
Sr. No. |
Peaks Observed in IR Spectra of drug+ polymer |
Functional Group |
|
1 |
3901.99 - 3602.52 |
O-H or N-H stretching (broad; H-bonded groups) |
|
2 |
3331.07 - 3207.62 |
N-H stretching (amines or amides) |
|
3 |
2960.73 - 2852.72 |
C-H stretching (alkanes, possibly polymer chains) |
|
4 |
2281.79 - 2112.05 |
C≡N or C≡C stretching (triple bonds) |
|
5 |
1649.14 |
C=O stretching (carbonyl) / C=N stretching (imine) |
|
6 |
1571.99 |
C=C stretching (aromatic groups) |
|
7 |
1402.25 |
C-H bending (alkane or aromatic C-H) |
|
8 |
1340.53 |
C-N stretching (amines) |
|
9 |
1247.81 |
C-O stretching (ethers/alcohols) |
9.4.2 Comparison showing the common peaks between: Lamivudine (pure drug) spectrum and Drug + Polymer spectrum.
|
Sr. No. |
Peaks Observed in IR Spectra of lamivudine (pure drug) and drug+ polymer. |
Common functional group present. |
|
1 |
3901.99 |
O-H/N-H stretching (broad) |
|
2 |
3331.07 |
N-H stretching |
|
3 |
2281.79 |
C≡N or C≡C stretching |
|
4 |
2112.05 |
C≡N or C≡C stretching (extension) |
|
5 |
1649.14 |
C=O or C=N stretching |
|
6 |
1571.99 |
C=C stretching (aromatic) |
|
7 |
1402.25 |
C-H bending (alkane/aromatic) |
|
8 |
1340.53 |
C-N stretching |
Conclusion: The comparison of IR spectra of Lamivudine (pure drug) and the drug–polymer mixture revealed that all major characteristic peaks of Lamivudine were retained in the drug–polymer spectrum. The presence of common peaks corresponding to functional groups such as O-H/N-H stretching, C≡N stretching, C=O/C=N stretching, and aromatic C=C stretching indicates that there was no significant chemical interaction between the drug and the polymer. This suggests that Lamivudine remains chemically stable in the presence of the polymer, ensuring the integrity of the drug during formulation.
9.5 Precompression studies of Tablet Blend:
Table No.15: Micromeritics study.
|
Formulation |
Bulk density (g/cm3) |
Tapped density (g/cm3) |
Hausner ratio |
% Compressibility |
Angle of repose (θ) |
|
F1 |
0.55 |
0.74 |
1.33 |
25 |
20.30 ? |
|
F2 |
0.53 |
0.71 |
1.36 |
25.35 |
21.80 ? |
|
F3 |
0.52 |
0.69 |
1.32 |
24.82 |
23.26 ? |
|
F4 |
0.49 |
0.67 |
1.36 |
25.35 |
24.22 ? |
|
F5 |
0.55 |
0.70 |
1.27 |
21.67 |
22.29 ? |
|
F6 |
0.57 |
0.72 |
1.26 |
21.42 |
19.29 ? |
|
F7 |
0.53 |
0.69 |
1.30 |
24.37 |
18.26 ? |
|
F8 |
0.53 |
0.75 |
1.41 |
26.33 |
25.17 ? |
The result of angle of repose is less than 30 indicates good flow properties of immediate release powder mix. This was further supported by lower Carr′s index value. This all formulations was
found to exhibit good to excellent flow properties.
9.5.1 post-compression study of Lamivudine tablets:
Table No.16: Post-compression study.
|
Sr. No. |
Formulation code |
Hardness (kg/cm2) |
Diameter (mm) |
Thickness (mm) |
Friability % |
Drug content (%) |
Weight Variation (mg) |
|
1. |
F1 |
4.75 |
9.51 |
4.81 |
0.47 |
97.50±1.31 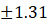
|
397-402 |
|
2. |
F2 |
5.65 |
9.50 |
5.220 |
0.32 |
96.46±1.12 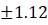
|
395-403 |
|
3. |
F3 |
5.05 |
9.51 |
4.85 |
0.49 |
82.76±1.0 
|
394-399 |
|
4. |
F4 |
4.85 |
9.50 |
5.03 |
0.32 |
94.60±1.46 
|
394-404 |
|
5. |
F5 |
5.95 |
9.50 |
5.47 |
0.71 |
97±1.45 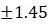
|
398-401 |
|
6. |
F6 |
6.25 |
9.51 |
5.18 |
0.74 |
89.86±1.13 
|
397-403 |
|
7. |
F7 |
6.15 |
9.50 |
5.17 |
0.56 |
86.38±1.31 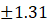
|
395-401 |
|
8. |
F8 |
5.35 |
9.50 |
5.01 |
0.61 |
87.14±1.14 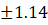
|
396-402 |
9.5.2 Cumulative % drug release of lamivudine floating tablets:
The release rate of lamivudine loaded floating tablets was determined using USP dissolution testing apparatus type Π (paddle type). The dissolution test was performed using 900 ml of0.1 N HCL at 37±
Table No.17.
Table No.17: In-vitro % drug release profile of batch F1-F8.
|
Time (hrs) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
|
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
1 |
11.31 |
8.72 |
64 |
22.72 |
19.26 |
20.30 |
86.63 |
42.58 |
|
2 |
24.62 |
52.26 |
87.67 |
43.10 |
40.68 |
43.79 |
99.59 |
57.09 |
|
3 |
32.74 |
64.35 |
94.75 |
55.19 |
54.16 |
52.95 |
107.53 |
65.04 |
|
4 |
42.24 |
76.61 |
98.90 |
69.18 |
62.10 |
65.73 |
116.17 |
79.89 |
|
5 |
53.46 |
86.29 |
99.24 |
70.22 |
66.77 |
69.53 |
120.83 |
86.63 |
|
6 |
57.96 |
95.96 |
112.20 |
84.73 |
74.71 |
86.80 |
138.28 |
93.37 |
|
7 |
63.46 |
94.06 |
148.30 |
89.40 |
77.82 |
89.57 |
133.10 |
101.31 |
|
8 |
69.36 |
104.94 |
99.07 |
99.24 |
84.04 |
93.89 |
137.25 |
107.71 |
|
9 |
78.17 |
105.81 |
111.68 |
99.59 |
87.15 |
100.80 |
134.83 |
120.49 |
|
10 |
91.99 |
113.93 |
117.38 |
120.66 |
102.01 |
114.10 |
134.65 |
121.87 |
|
11 |
99.24 |
121.01 |
107.88 |
119.28 |
109.78 |
117.55 |
139.49 |
124.98 |
|
12 |
103.56 |
125.15 |
106.50 |
121.70 |
119.28 |
118.24 |
141.22 |
126.36 |
Cumulative % Drug Release of Tablet:

Figure No.16: Cumulative drug release of tablets.
Prepared tablet of lamivudine and HPMC K 100 M of batch F1 show 99.24% of drug release in 11 hr and F5 batch of polymer used Xanthum gum show 87.15% drug release in 9 hr.
Floating Lag Time of Tablets of Prepared Batch.
Table No.18: Floating Lag Time.
|
Formulation |
Floating lag time (sec) |
Total floating time (hr) |
|
F1 |
98 |
>12 |
|
F2 |
5 |
>12 |
|
F3 |
22 |
>12 |
|
F4 |
133 |
>12 |
|
F5 |
107 |
>12 |
|
F6 |
64 |
>12 |
|
F7 |
36 |
>12 |
|
F8 |
51 |
>12 |
9.6 Kinetics of drug Release
Table No.19: Drug Release Kinetics of Formulations F1 To F8.
|
Formulation code |
First order plot (R2) |
Zero order plot (R2) |
Higuchi plots (R2) |
Hixon-Crowel cube root plot (R2) |
Korsmeyer-peppas plots |
Best fit model |
|
|
N |
R2 |
||||||
|
F1 |
0.7554 |
0.9850 |
0.9527 |
0.9028 |
0.8508 |
0.9895 |
Korsmeyer-peppas |
|
F2 |
0.8836 |
0.8783 |
0.9604 |
0.9400 |
0.9895 |
0.8695 |
Korsmeyer-peppas |
|
F3 |
0.5648 |
0.4463 |
0.6840 |
0.6984 |
0.2095 |
0.9895 |
Korsmeyer-peppas |
|
F4 |
0.8466 |
0.9466 |
0.9842 |
0.8855 |
0.6523 |
0.9895 |
Korsmeyer-peppas |
|
F5 |
0.8364 |
0.9394 |
0.9718 |
0.7868 |
0.6499 |
0.9896 |
Korsmeyer-peppas |
|
F6 |
0.8646 |
0.9430 |
0.9855 |
0.8904 |
0.6725 |
0.9891 |
Korsmeyer-peppas |
|
F7 |
0.1743 |
0.2149 |
0.1315 |
0.3461 |
1.1031 |
0.9517 |
Korsmeyer-peppas |
|
F8 |
0.8216 |
0.8984 |
0.9942 |
0.9267 |
0.4594 |
0.9318 |
Higuchi plots(R2) |
Observation:
Conclusion:
The drug release kinetics study indicated that the release mechanism of lamivudine microspheres (F1–F7) predominantly followed the Korsmeyer-Peppas model, suggesting a combination of diffusion and erosion mechanisms. Formulation F8 followed the Higuchi model, indicating a diffusion-controlled release. These findings suggest that the formulations exhibit controlled drug release behavior suitable for sustained drug delivery.
9.7 Accelerated stability studies:
Stability studies are the most vital and important part foe improving the life of pharmaceutical dosage form. The tablets are tested by wrapping them in aluminum foil. These tablets were kept in incubator and then were after 30 days and analyzed for the physical characterization, visual defect, hardness, friability, thickness, dissolution tests, disintegration time. There was no change in the percentage release of lamivudine from the formulations stored at different temperature for 30 days. It showed that all the formulation are physically stable. There was no change in the formulation the result was presented in Table No.20, Table No.21, Table No.22.
Table No.20: Physicochemical Data of Selected Formulation Before and After Stability Study.
|
Formulation |
Hardness (kg/cm2) |
Thickness (mm) |
Diameter (mm) |
|||
|
|
Before |
After |
Before |
After |
Before |
After |
|
F1 |
4.75 |
4.75 |
4.81 |
4.81 |
9.51 |
9.50 |
|
F5 |
5.95 |
5.95 |
5.47 |
5.47 |
9.50 |
9.50 |
Table No.21: Physicochemical data of selected formulation before and after stability study.
|
Formulation |
Friability % |
Weight variation (mg) |
Floating lag time (sec) |
|||
|
|
Before |
After |
Before |
After |
Before |
After |
|
F1 |
0.47 |
0.53 |
Passes |
Passes |
98 |
94 |
|
F5 |
0.71 |
0.78 |
Passes |
Passes |
107 |
105 |
Table No.22: Physicochemical Data of Selected Formulation Before and After Stability Study.
|
Time (hr) |
Before stability |
After stability |
Before stability |
After stability |
|
|
Cumulative % drug release |
Cumulative % drug release |
Cumulative % drug release |
Cumulative % drug release |
|
|
Formulation batch F1 |
Formulation batch F5 |
||
|
1 |
11.31 |
10.80 |
19.26 |
18.40 |
|
2 |
24.62 |
23.58 |
40.68 |
40.16 |
|
3 |
32.74 |
31.70 |
54.16 |
53.64 |
|
4 |
42.24 |
41.55 |
62.10 |
61.41 |
|
5 |
53.46 |
52.60 |
66.77 |
66.42 |
|
6 |
57.96 |
57.44 |
74.71 |
73.50 |
|
7 |
63.46 |
67.80 |
77.82 |
76.79 |
|
8 |
69.36 |
77.48 |
84.04 |
83.69 |
|
9 |
78.17 |
91.12 |
87.15 |
86.98 |
|
10 |
91.99 |
98.55 |
102.01 |
101.49 |
|
11 |
99.24 |
100.80 |
109.78 |
109.09 |
|
12 |
103.56 |
102.70 |
119.28 |
118.07 |
Cumulative % Drug Release Before Stability:

Figure No.17: Cumulative % drug release before stability study.
Cumulative % Drug Release After Stability:

Figure No.18: Cumulative % Drug Release After Stability Study.
SUMMARY AND CONCLUSION
Summary:
This study aimed to develop lamivudine-loaded spray-dried microspheres and gastroretentive floating tablets using various polymers. Lamivudine was characterized as a white, odourless, bitter drug with a melting point of 160–161°C, soluble in water and sparingly soluble in alcohols. A UV spectrophotometric calibration curve in 0.1N HCl exhibited excellent linearity (R² = 0.9992). Microspheres were prepared using Eudragit L100, HPMC, and ethyl cellulose in various ratios. The highest yield (23%) was observed in batch F10, while most batches showed low yields, indicating the need for process optimization. FTIR analysis confirmed no chemical interaction between lamivudine and polymers, establishing formulation compatibility.
Tablet blends exhibited good to excellent flow properties. Compressed tablets passed physical evaluations including hardness, friability, and uniformity. In vitro dissolution studies revealed that batch F1 (with HPMC K100M) achieved 99.24% drug release over 11 hours, while batch F5 (with xanthan gum) released 87.15% in 9 hours. All tablets displayed a floating lag time <2 minutes and sustained buoyancy >12 hours. Drug release kinetics indicated that most formulations followed the Korsmeyer-Peppas model, suggesting a combination of diffusion and erosion mechanisms. Formulation F8 followed the Higuchi model, pointing to a diffusion-dominant release.
CONCLUSION:
REFERENCES


Satyam Pendor*, Dr. A. V. Chandewar, Dr. M. A. Channawar, Gaurav Magar, Design, Development and Optimization of Floating Gastro-Retentive Drug Delivery System, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 4625-4643. https://doi.org/10.5281/zenodo.15532437
 10.5281/zenodo.15532437
10.5281/zenodo.15532437
