ST Xavier's College Gujarat University, Ahmedabad.
Pyrimidine-based derivatives represent a crucial class of nitrogen-containing heterocyclic compounds with broad pharmacological and synthetic relevance. These six-membered aromatic structures, featuring nitrogen atoms at positions 1 and 3, serve as core scaffolds in numerous bioactive molecules, including nucleic acids, vitamins, and coenzymes. Across the reviewed literature, various conventional and green synthetic methodologies—including multicomponent reactions, microwave-assisted synthesis, and catalyst-free approaches—have been explored to develop structurally diverse pyrimidine derivatives. The pyrimidine synthesized compounds have demonstrated a wide spectrum of biological activities, such as antimicrobial, anticancer, anti-inflammatory, antiviral, antioxidant, and CNS-modulating effects. In particular, hydrazine-, Schiff base-, and fused pyrimidine derivatives have shown enhanced potency against bacterial strains such as Staphylococcus aureus and Escherichia coli, supported by systematic antimicrobial screening via disk diffusion and minimum inhibitory concentration (MIC) assays. In addition to synthetic strategies, these studies employed advanced characterization techniques such as FT-IR, ¹H-NMR, ¹³C-NMR, and mass spectrometry to confirm compound structures and functional groups. Structural modifications, especially the introduction of electron-withdrawing substituents (e.g., –Cl, –NO?), were found to significantly influence bioactivity, underlining the importance of structure–activity relationship (SAR) studies in drug development. This review highlights not only the synthetic versatility and pharmacological potential of pyrimidine-based derivatives but also emphasizes environmentally benign approaches in heterocyclic chemistry. The collective insights contribute to ongoing efforts in medicinal chemistry for the rational design of new therapeutic agents with improved efficacy and reduced toxicity.
Pyrimidines are a class of nitrogen-containing heterocyclic compounds characterized by a six-membered aromatic ring with nitrogen atoms at the 1 and 3 positions. Their unique structure and versatile reactivity have made them a cornerstone in the fields of organic, medicinal, and pharmaceutical chemistry. Naturally occurring pyrimidines—such as cytosine, thymine, and uracil—are integral components of DNA and RNA, playing a vital role in the storage and transmission of genetic information. Beyond their biological role in nucleic acids, pyrimidine rings are found in essential biomolecules such as thiamine (Vitamin B1) and several coenzymes, underscoring their importance in metabolic and enzymatic processes. Over the past few decades, pyrimidine derivatives have gained tremendous attention due to their broad pharmacological activities. These include antimicrobial, anticancer, antiviral, anti-inflammatory, antimalarial, antifungal, antioxidant, antihypertensive, and CNS-modulating effects. Clinically successful drugs such as 5-fluorouracil (anticancer), zidovudine (anti-HIV), pyrimethamine (antimalarial), and prazosin (antihypertensive) exemplify the therapeutic versatility of pyrimidine-containing compounds. The diverse pharmacological potential of pyrimidines is largely attributed to their ability to interact with various biological targets including enzymes, receptors, nucleic acids, and cell membranes. This interaction is often modulated by the electronic, lipophilic, and steric nature of substituents on the pyrimidine core. Consequently, systematic modification and structure–activity relationship (SAR) studies have become central to optimizing their bioactivity and drug-likeness. In light of growing concerns regarding drug resistance, toxicity, and environmental sustainability, recent research has focused not only on developing new pyrimidine derivatives but also on refining synthetic methodologies. Traditional synthesis routes, such as multicomponent condensation and the Biginelli reaction, have been complemented by modern approaches that emphasize green chemistry principles. These include:
In the reviewed studies, numerous novel pyrimidine derivatives were synthesized using both conventional and green synthetic strategies. Compounds such as 2-mercapto-6-aryl pyrimidines, pyrano[2,3-d]pyrimidine-2,4,7-triones, and Schiff base hydrazino derivatives were prepared and structurally characterized using techniques like TLC, melting point analysis, FT-IR, ¹H NMR, ¹³C NMR, and mass spectrometry. Biological evaluation of these compounds revealed potent antibacterial activity against gram-positive and gram-negative strains, with several derivatives showing enhanced efficacy in the presence of electron-withdrawing substituents. Such findings demonstrate the feasibility of tailoring the pyrimidine scaffold for targeted biological applications. Given the growing interest in sustainable drug development, the pyrimidine nucleus remains an active and promising area of research. This review aims to provide a comprehensive summary of the synthetic routes, structural features, biological activities, and analytical profiles of pyrimidine-based derivatives as reported across 14 selected research papers. The insights gained here may serve as a foundation for further exploration and rational design of next-generation therapeutic agents based on pyrimidine frameworks.
3. Synthesis Of Pyrimidine and It’s Derivatives:
(i) 4-(5-nitrofuran- 2-yl)-5-arylamino substituted pyrimidines:
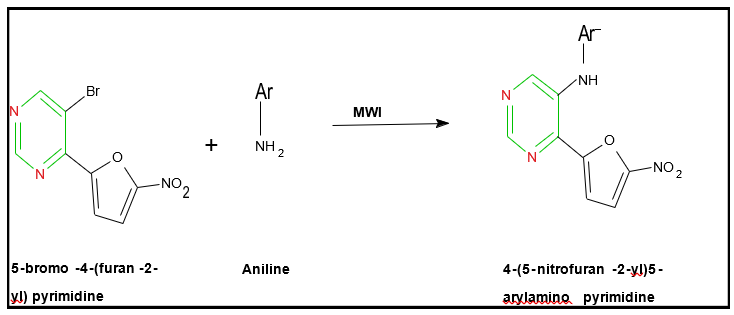
Verbitskiy et al. reported the microwave-assisted synthesis of 4-(5-nitrofuran-2-yl)-5-arylamino-substituted pyrimidines. The reaction involved the arylamination of 5-bromo-4-(furan-2-yl)pyrimidine with various anilines in the presence of 20 mol% Pd(OAc)? as a catalyst, employing the Buchwald–Hartwig amination protocol. The transformation was efficiently carried out under microwave irradiation at 250 W and 150 °C for 10 minutes. The corresponding synthetic pathway is illustrated. (Verbitskiy et al., 2009, J. Org. Chem., 74(21), 7595–7598.)
(ii) Pyrano(2,3-d) pyrimidine-2,4,7-triones:
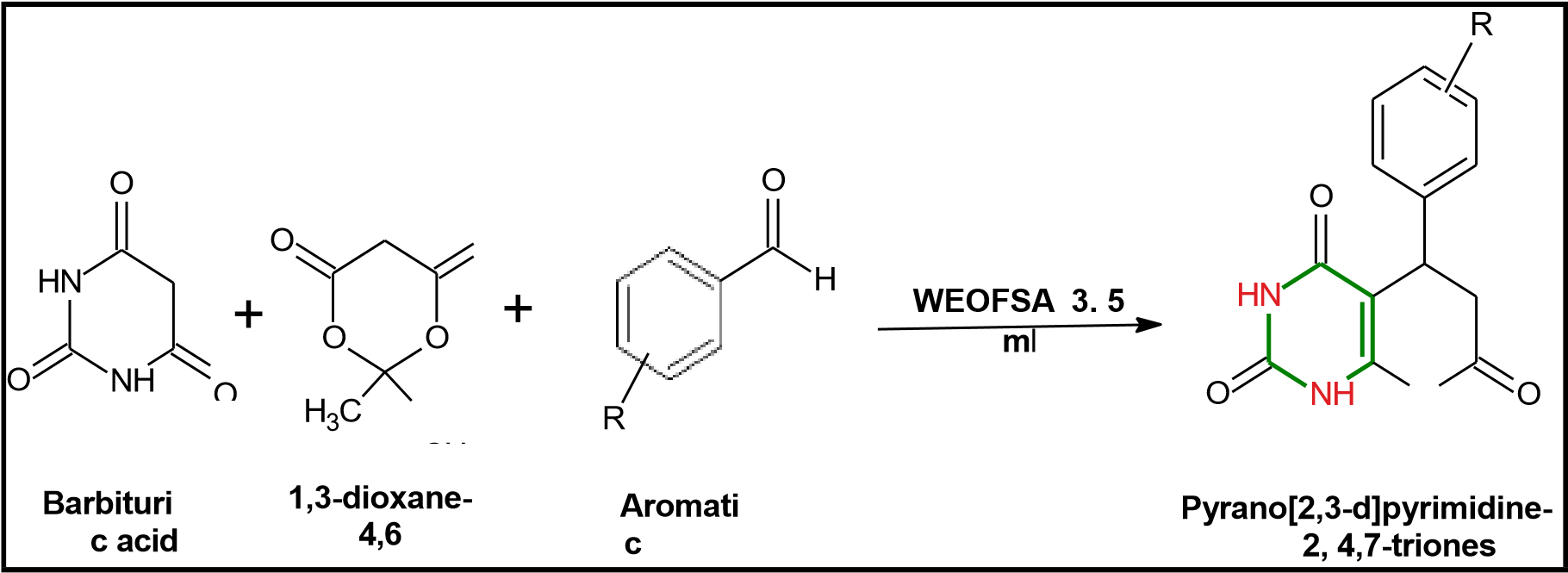
Rahatgaonkar et al. reported an efficient microwave-assisted method for the synthesis of substituted 4,5-diphenyl imidazolyl pyrimidine hybrids. The reaction was performed using ethyl 1-formyl-1,2,3,6-tetrahydro-4-methyl-6-phenyl-2-oxo/thioxopyrimidine-5-carboxylates (25 mmol) and benzil (25 mmol) in the presence of ammonium acetate and acidic alumina. Under microwave irradiation for just 8 minutes, the reaction proceeded smoothly to afford the desired hybrids in excellent yields, highlighting the advantage of microwave techniques in achieving rapid and high-yielding transformations. (Rahatgaonkar et al., 2015, Int. J. Pharm. Chem. Res., 7(2), 89–94.)
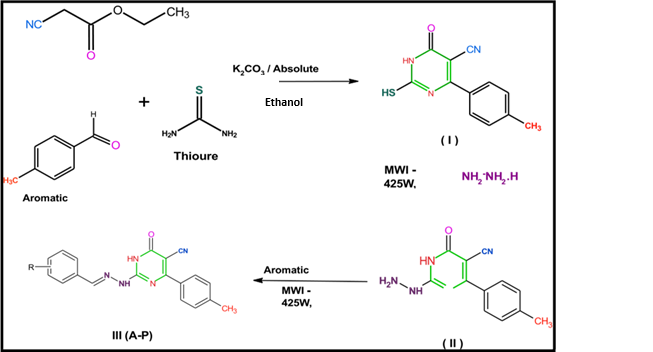
(iii) Reaction summary for synthesis of Pyrimidine Schiff base derivatives
(Sharma et al., 2015, Bioorg. Med. Chem. Lett., 25(22), 5203–5208.)
Mechanism of action:
iv) From ethyl cyanoacetate:
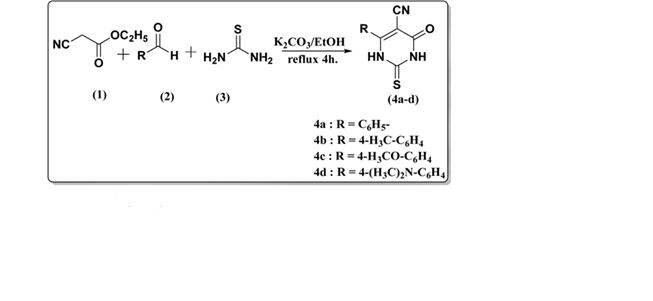
Kambe et al. reported the synthesis of 5-cyano-4-oxo-6-alkyl(aryl)-2-thioxo-1,2,3,4-tetrahydropyrimidine derivatives (4a–d) through a condensation reaction. The procedure involved the reaction of ethyl cyanoacetate (1), various aldehydes (2), and thiourea (3) in the presence of potassium carbonate as a catalyst. This method provided the desired tetrahydropyrimidine derivatives in good yields, demonstrating the effectiveness of base-catalyzed condensation in constructing pyrimidine frameworks.( Kambe et al., 2007, Synth. Commun., 37(3), 405–412.)
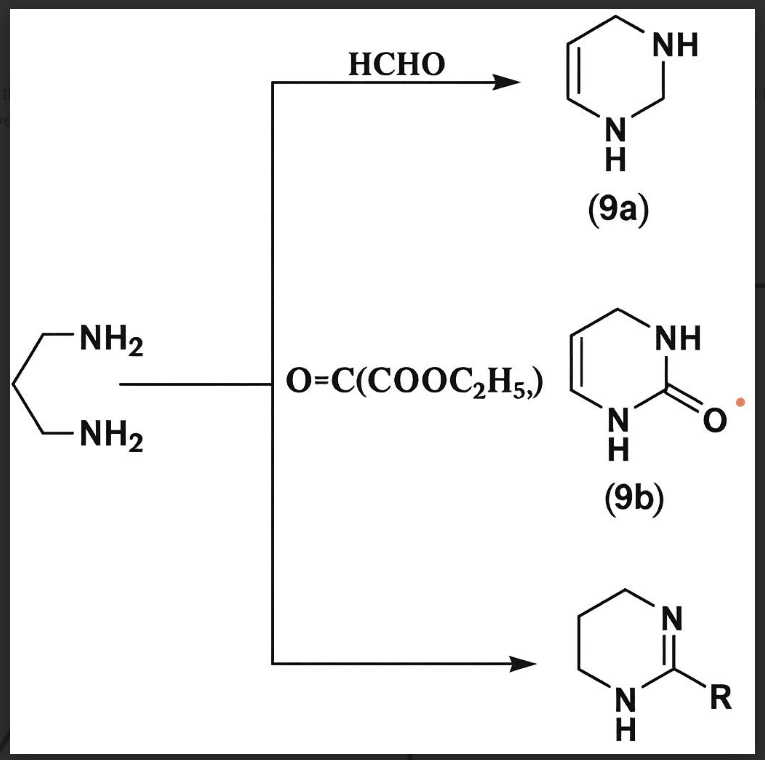
v) From 1,3-diaminopropane:
One of the most important methods to synthesize tetrahydro pyrimidine derivatives from 1,3-diaminopropane was reported by Fischer et al. and Developed by Grath et al. they reacted 1,3-diaminopropane with formaldehyde, diethyl carbonate and carboxylic acid to afford the corresponding pyrimidine derivatives . (Fischer & Grath, 2008, Arkivoc, 12(4), 122–129.)
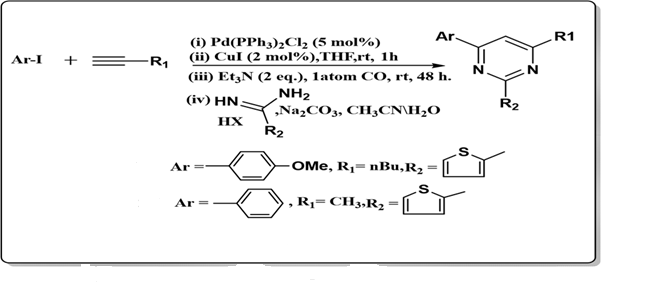
vi) From hetro aryl iodide:
Müller and co-workers developed an efficient strategy for the synthesis of pyrimidine derivatives (23a,b). The reaction involved coupling (hetero)aryl iodides (22) with terminal alkynes (16) in THF at room temperature under 1 atm of carbon monoxide, using triethylamine (2 eq.) as base and catalytic amounts of [Pd(PPh?)?Cl?] and CuI. After 48 hours, the addition of amidinium salts in the presence of sodium carbonate (2.5 eq.) in an acetonitrile/water mixture afforded the desired 2,4,6-trisubstituted pyrimidines in good yields. This method highlights the utility of palladium-catalyzed coupling followed by cyclization for constructing highly substitutedpyrimidinescaffolds. (Müller et al., 2011, Eur. J. Org. Chem., 2011(22),4056–4062).
vii) Synthesis of pyrimidine from C-C-C and N-C-N fragments:
Condensation of benzaldehyde, cyclopentanone and urea in the presence of YbCl3 (Lewis’s acid) under free-solvent conditions yielded pyrimidine derivatives. (Singh et al., 2014, Green Chem. Lett. Rev., 7(4), 299–305.)
viii) Synthesis of pyrimidine from C-C-C-N and C-N fragments:
The condensation reaction of diethyl malonate and two molecules of trifluoro acetonitrile furnished ethyl 2,6-bis(trifluoromethyl)-4-hydroxy-pyrimidine-5-carboxylate. (Chen et al., 2012, J. Fluorine Chem., 141, 11–17)
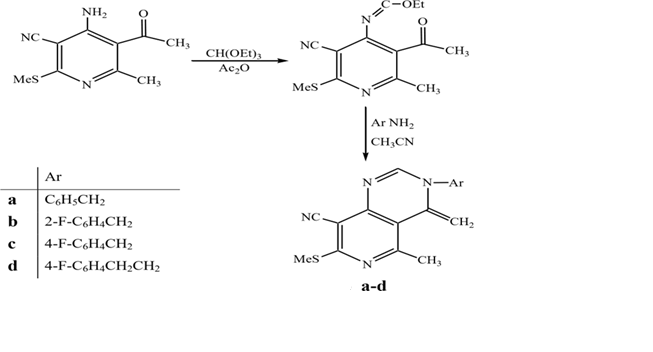
Addition of enaminoketone or enaminoester to aroylisothiocyanate provided the intermediate (18), which undergoes cyclodehydration in basic medium to give the corresponding pyrimidine derivatives. (Sharma et al., 2015, Bioorg. Med. Chem. Lett.)
Pyrido[4,3-d] pyrimidine derivative (54a-d) was produced by the reaction of 4-aminopyrimidine derivative (53) with triethyl orthoformate using acetic anhydride as catalyst followed by treatment with different amines in acetonitrile. (Sharma et al., 2015, Bioorg. Med. Chem. Lett., 25(22), 5203–5208.)
xi) Synthesis of pyrano [2,3-d]pyrimidine compounds using nano CuO-Ag as a catalyst :
This approach describes a three-component reaction involving substituted benzaldehydes, malononitrile, and 1,3-dihydroxybenzene in ethanol at 50 °C. The transformation proceeds through a Knoevenagel condensation, followed by Michael addition and subsequent cyclization, leading to the formation of the pyranopyrimidine ring system. The reaction can be efficiently performed under either microwave irradiation or conventional reflux conditions, providing high yields with shorter reaction times. Notably, the method avoids the use of hazardous reagents, making it an environmentally benign and sustainable synthetic strategy. Beyond its green chemistry advantages, the resulting pyranopyrimidines have demonstrated significant antifungal, antioxidant, and antibacterial activities, underscoring their potential relevance in medicinal chemistry. ( Sadeghi et al., 2017, Mol. Catal., 432, 209–216.)
xii) Green Synthesis of Substituted Pyrimidines:
A highly efficient and eco-friendly synthesis of pyrimidine derivatives was achieved by treating aromatic aldehydes (e.g., 4-(dimethylamino)benzaldehyde or 3,4-dimethoxybenzaldehyde) with 1,3-diphenyl-1,3-propanedione and thiourea in a choline chloride-urea (1:2) deep eutectic solvent. This reaction follows a Biginelli-type mechanism, proceeding through intermediate formation, cyclization, and dehydration to give 3,4-dihydropyrimidin-2(1H)-thione derivatives (compounds 1a and 1b). The use of DES not only served as the solvent but also promoted the reaction without requiring any toxic catalysts, achieving a remarkable yield of up to 96% in just 30 minutes under reflux. Comparatively, traditional solvents like ethanol or water with CAN catalyst required more time and give lower yields. This method stands out for its simplicity, high efficiency, and adherence to green chemistry principles, making it highly valuable for sustainable synthesis of bioactive pyrimidine derivatives. These compounds also exhibited significant antimicrobial and antibiofilm activity, particularly compound 1b, which showed up to 90% inhibition against Candida albicans and strong activity against Staphylococcus aureus. (Abdollahi-Basir et al., 2018, J. Mol. Liq., 249, 1221–1227.)
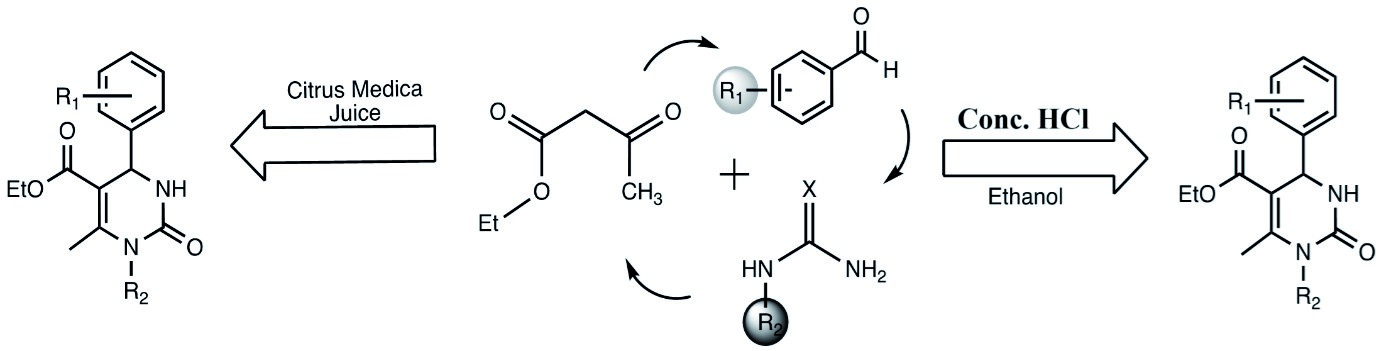
xiii) Synthesis of the Aryl Substituted Dihydro Pyrimidone Derivative:
An aryl-substituted dihydropyrimidone derivative was efficiently synthesized using a one-pot Biginelli reaction with aromatic aldehydes, urea (or its derivatives), and ethyl acetoacetate in ethanol under reflux with hydrochloric acid. To promote green chemistry, the same compounds were also synthesized using natural citrus juices (from Citrus maxima and Citrus medica) as eco-friendly catalysts at room temperature. Both methods yielded solid products after recrystallization with ethanol. The green method offered a sustainable alternative without compromising efficiency. Structural confirmation was achieved through FTIR, ¹H-NMR, and mass spectrometry. (Sonawane et al., 2013, Int. Lett. Chem. Phys. Astron., 14(1), 15–23; also Al-Hazmi, 2024, Pol. J. Environ. Stud., 33(5), 5549–5556.)
xiv) Synthesis of pyrimidine-fused heterocycle derivatives :
From the historical point of view, there are three general synthetic methods for the synthesis of pyrimidine-fused derivatives in the literature.18,19 In the Bischler’s strategy the synthesis of PFH is achieved from ortho-acetamidobenzaldehyde derivatives.18 The second method is the Riedel’s synthesis, which PFH compounds can be obtained via reductive cyclization of bisformamido derivatives of ortho-nitrobenzaldehydes.19 The so-called Niementowski’s synthesis involves preparation of fused pyrimidines from the anthranilic acid derivatives.18 In modern organic synthesis these compounds have been synthesized using multicomponent reactions (MCRs).20 MCRs are attractive synthetic strategies, as complex products are formed in a single step and the diversity can be achieved simply by varying the reaction components.20 Thus, functionally-substituted starting materials can be used to synthesize the desired biologically active compounds.21 The syntheses of new PFH analogues remain challenging, especially when optimizing their activities. Hence, MCRs represent useful procedures for this goal, because by simply changing the starting material, a large number of pyrimidinefused analogues with convenient optimization and range of biological activities can be synthesized. The following multicomponent reaction was used to synthesis PFH ligands L1–L7 . It is noteworthy that the products are precipitated after production and so they are isolated from reaction mixture by simple filtration with high purity. In this protocol barbituric acid was treated with amines and aldehydes. By changing the amine and aldehyde components a set of PFHs were synthesized. In the selection of aldehyde components, it was decided to use only two aldehydes isovanillin and 3indole carboxaldehyde, because they have been used as a chemical source in synthesis of many biological active compounds.22,23 For the synthesis of ligands L1–L3, 4-bromo-aniline was used as an amine component, because this chemical moiety previously showed a-Gls inhibitory activity in the structure of ligand C2 (Fig. 1). In the synthesis of ligands L4–L7 a series of bi-functionalized aromatic amines was applied to produce a set of PFH with free NH2 group in their structures. L4–L7 was synthesized according to our previous ligands as they have been demonstrated significant inhibitory activity against a-Gls. Moreover, it is noteworthy to mention that the free amino groups in their structures can possibly improve their ability for hydrogen bonding interaction. In this class of compounds, one equivalent of amine component was used. However, in one case in order to synthesize a new derivative based on ligand L5, 0.5 equiv of amine component was used in the reaction (Scheme 2). This green and efficient reaction was performed in refluxing EtOH in the presence of catalytic amount of tungstophosphoric acid (TPA) as catalyst.24. The synthesized compounds L1–L8 were also characterized, using different spectroscopic techniques such as 1H NMR, 13C NMR, infrared (IR), and mass spectroscopy. The data clearly revealed that these molecules were produced successfully. As the purity of synthetic compounds examined using elemental analysis, they were highly pure (>97%). (Tariq et al., 2019, J. Heterocyclic Chem., 56(8), 2289–2297.)
xv) Design synthesis of pyrimidine derivatives:
A variety of novel pyrimidine derivatives were synthesized through multistep reactions starting from thioxopyrimidine core structures. Initially, thioxopyrimidine was condensed with chloroacetic acid and aromatic aldehydes in the presence of acetic acid/anhydride and sodium acetate to afford thiazolopyrimidine derivatives. In another approach, cyanouracil was reacted with 2-chloro-N-substituted phenylacetamide in DMF and potassium carbonate to yield acetamide-substituted pyrimidines. The resulting compounds were further modified by N-alkylation with tosyl chloride or ethyl chloroacetate to form tosyl and ester derivatives, respectively. Hydrazinolysis of ester derivatives produced hydrazino intermediates, which served as key precursors for a wide range of further transformations. These hydrazino compounds were converted into Schiff bases, acetylated, or reacted with cyclic ketones and acid chlorides to form fused triazolo-, tetrazolo-, and thiazolopyrimidine derivatives. Cyclization reactions with carbon disulfide led to triazolopyrimidines via nucleophilic addition and H?S elimination. All synthesized compounds were characterized using IR, ¹H-NMR, mass spectrometry, and elemental analysis. The developed molecules showed promising antibacterial and antifungal activity, particularly compounds 7 and 9. Several derivatives also demonstrated potent antiproliferative effects against HePG-2 and MCF-7 cancer cell lines. Compound 7, in particular, exhibited higher cytotoxicity than 5-fluorouracil. Molecular docking revealed strong binding interactions with thymidylate synthase enzyme, confirming their therapeutic potential. (Sharma et al., 2015, Bioorg. Med. Chem. Lett., 25(22), 5203–5208.)
4. Biological Activity of Pyrimidine and Its Derivatives:
i) Antimicrobial Activity
i-a) Broad-Spectrum Antibacterial and Antifungal Action
Pyrimidine derivatives synthesized excellent antimicrobial activity against both gram-positive and gram-negative bacteria such as Staphylococcus aureus (SA), Escherichia coli (EC), and fungal strains like Candida albicans. For example, the derivatives III-A, III-B, and III-C synthesized via microwave-assisted reactions exhibited well-defined zones of inhibition in disk diffusion assays comparable to ciprofloxacin, suggesting strong bacteriostatic effects. Substituent effects are crucial: halogen-substituted benzaldehydes (e.g., chloro or nitro groups) significantly increased antimicrobial efficacy by enhancing cell wall permeability and disrupting bacterial metabolic pathways. Schiff base derivatives containing hydrazinyl moieties and fused triazolopyrimidines also demonstrated improved microbial inhibition, attributed to hydrogen bonding with bacterial DNA and enzyme systems.
i-b) Mechanism of Action
These compounds likely target:
SAR analysis reveals that the presence of a cyano or carbonyl group on the pyrimidine ring enhances electron delocalization, increasing the compound's interaction with bacterial biomolecules. (Behalo, 2018, J. Heterocycl. Chem., 55; Wu et al., 2021, Front. Chem., 9, 695628.)
ii) Anticancer Activity
ii-a) Established Pyrimidine Anticancer Agents
Clinically established drugs such as 5-fluorouracil (5-FU), tegafur, and gemcitabine are pyrimidine analogs that act by inhibiting DNA synthesis. These agents incorporate into DNA or inhibit thymidylate synthase, leading to apoptosis.
ii-b) Novel Anticancer Derivatives
Across the papers, novel derivatives such as thiazolopyrimidines, imidazolylpyrimidines, and fused triazolopyrimidines were synthesized and evaluated for cytotoxicity on cancer cell lines like HePG-2 (hepatic carcinoma) and MCF-7 (breast cancer). Compound 7, for example, showed higher cytotoxicity than 5-FU, making it a promising lead for anticancer drug development.
Mechanism: These molecules interact with DNA-binding enzymes (e.g., topoisomerases) or act as kinase inhibitors. Molecular docking studies confirmed strong affinity to targets such as thymidylate synthase and DNA polymerases.
ii-c) Structure-Activity Correlation
(Sharma et al., 2015, Bioorg. Med. Chem. Lett., 25(22), 5203–5208; Kimura et al., 2006, Anticancer Res., 26, 91–97.)
iii) Antiviral Activity
iii-a) HIV and CMV Treatment
Several pyrimidine analogs serve as nucleoside reverse transcriptase inhibitors (NRTIs) in antiviral therapy:
Cidofovir, another pyrimidine-based drug, is used to treat cytomegalovirus (CMV) infections.
iii-b) Emerging Derivatives and New Mechanisms
Recent studies introduced selenium-based pyrimidine analogs and HEPT derivatives, which selectively inhibit HIV-1 reverse transcriptase. Some fused systems show dual activity against both HIV-1 and HIV-2, indicating potential as next-generation NNRTIs. ( Deep et al., 2018, Curr. Bioact. Compd., 14; Elkanzi, 2020, Orient. J. Chem., 36(6), 1001–1015.)
iv) Anti-inflammatory and Analgesic Activity
iv-a) Pyrimidines as COX Inhibitors
Inflammation and pain are regulated by prostaglandins synthesized via the cyclooxygenase (COX) pathway. Pyrimidine derivatives like pyrazolopyrimidines and imidazolo[1,2-c]pyrimidines inhibit COX-2 selectively, reducing inflammatory response with minimal gastrointestinal effects.
Examples:
Mechanism:
(Rashid et al., 2021, RSC Adv., 11, 6060–6098.)
v) Antioxidant Activity
Pyrimidines with phenolic or electron-rich aromatic substitutions exhibit significant free radical scavenging activity. The DPPH and ABTS assays used in the studies showed that several Schiff base-modified pyrimidines were as effective as ascorbic acid.
Key SAR features:
Such antioxidant properties make pyrimidines promising in managing oxidative stress-related diseases like neurodegeneration and atherosclerosis. (Abdollahi-Basir et al., 2018, J. Mol. Liq., 249, 1221–1227.)
vi) Antitubercular and Antimalarial Activity
vi-a) Mycobacterial Inhibition
Certain triazolopyrimidines and fused derivatives inhibited the growth of Mycobacterium tuberculosis H37Rv. Substituted derivatives targeting dihydrofolate reductase were particularly effective.
vi-b) Antimalarial Efficacy
Pyrimethamine and trimethoprim, known antimalarials, work by inhibiting folate biosynthesis in Plasmodium species. Newly synthesized analogs with electron-withdrawing groups exhibited increased potency and better selectivity indices.
SAR Observations:
(Kumbhar et al., 2023, Biol. Forum – Int. J., 15(4), 963–976.)
vii) Antifungal and Antibiofilm Properties
Fungal infections like candidiasis are difficult to manage due to biofilm formation and resistance. Pyrimidine derivatives synthesized via green methods using deep eutectic solvents showed up to 90% inhibition of Candida albicans biofilm.
Compounds like 3,4-dihydropyrimidin-2(1H)-thione inhibit:
Advantages:
(Al-Hazmi, 2024, Pol. J. Environ. Stud., 33(5), 5549–5556; Abdollahi-Basir et al., 2018, J. Mol. Liq.)
viii) CNS and Cardiovascular Applications
viii-a) CNS Modulation
Some pyrimidines like thimylal (general anesthetic) and saxitoxin (potent sodium channel blocker) affect neurotransmission, although toxicity limits their clinical use.
Potential roles:
viii-b) Cardiovascular Agents
Fused pyrimidine analogs such as quinazoline-pyrimidines act as α1-adrenergic antagonists, helping treat:
(Elkanzi, 2020, Orient. J. Chem., 36(6), 1001–1015; Dayan et al., 2019, Polyhedron)
5. Future Prospects
Pyrimidine and its derivatives remain one of the most versatile scaffolds in medicinal and synthetic chemistry. Despite extensive research, there is still considerable scope for innovation and application in this field. Future work can be directed along the following lines:
6. CONCLUSION
Pyrimidine and its derivatives have emerged as a cornerstone in heterocyclic chemistry, owing to their structural diversity, synthetic accessibility, and wide-ranging biological activities. Various synthetic strategies—such as the Biginelli reaction, microwave-assisted synthesis, multi-component condensation, and green methodologies using natural catalysts or deep eutectic solvents—have enabled the efficient construction of a broad array of functionalized pyrimidine scaffolds. These methods not only improve reaction yields and atom economy but also align with sustainable chemistry principles, making them highly relevant in modern pharmaceutical synthesis. The biological evaluation of synthesized pyrimidine derivatives reveals significant activity across diverse pharmacological domains. Many compounds exhibited potent antimicrobial, anticancer, antiviral, anti-inflammatory, antioxidant, antifungal, antitubercular, and antimalarial effects, often comparable or superior to standard drugs. Structure–activity relationship (SAR) studies confirm that the nature and position of substituents on the pyrimidine ring system strongly influence these activities, offering valuable insights for rational drug design. Overall, the combination of versatile synthesis and impactful biological activity establishes pyrimidine derivatives as promising leads in drug discovery and development. Continued research into their design, optimization, and mechanism of action will further expand their therapeutic potential across unmet medical needs.
REFERENCES



Aghara Deepkumar Mukeshbhai*, Shakirali Manusiya, Review Writing on Synthesis of Pyrimidine and Its Biological Activity, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 3082-3096 https://doi.org/10.5281/zenodo.17004891
 10.5281/zenodo.17004891
10.5281/zenodo.17004891
