Valmik Naik College of Pharmacy, Telwadi, Kannad, Chh. Sambhajinagar
Drug design and discovery represent a multidisciplinary field combining chemistry, biology, pharmacology, and computational sciences to develop safe and effective therapeutic agents. This review provides an overview of the principles, methodologies, and current trends shaping modern drug development. The process begins with target identification and validation, followed by hit discovery, lead optimization, preclinical studies, and clinical trials. Approaches such as Quantitative Structure–Activity Relationship (QSAR), Computer-Aided Drug Design (CADD), pharmacophore modeling, and combinatorial chemistry play vital roles in predicting molecular interactions, optimizing lead compounds, and accelerating discovery timelines. Integration of artificial intelligence, machine learning, and high-throughput screening has revolutionized drug design, improved prediction accuracy and reducing costs. Natural products, synthetic chemistry, and biotechnology continue to be significant sources of novel therapeutics. Despite advancements, challenges such as high costs, long development periods, and high attrition rates persist. Future directions emphasize AI-driven modeling, precision medicine, sustainable chemistry, and nanotechnology-based delivery systems to enhance innovation and efficiency in pharmaceutical research.
Drug design and discovery form the cornerstone of pharmaceutical sciences, integrating chemistry, biology, pharmacology, and computational approaches to transform basic concepts into therapeutic agents. Over the past decades, several landmark drugs have demonstrated the power of rational discovery strategies. Statins, for instance, were first identified as fungal metabolites and subsequently optimized through synthetic modifications, revolutionizing the management of hypercholesterolemia and cardiovascular disease [12–16]. More recently, nutraceutical-based interventions have been evaluated as alternatives or adjuncts to conventional statin therapy, underscoring the continuous interplay between natural products, synthetic chemistry, and clinical practice in modern drug design [17].
The drug discovery process follows a series of well-established stages, including target identification, hit discovery, lead optimization, and preclinical development [1]. A key component of these stages is the use of Quantitative Structure–Activity Relationship (QSAR) models, which predict biological activity based on molecular descriptors and structural features [2]. Alongside QSAR, a wide range of computational and experimental tools—such as computer-aided drug design (CADD), chemical structure illustration, database searching, pharmacophore modeling, docking, and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) analysis— have become integral to the discovery pipeline [3,4]. These methodologies not only allow the visualization and prediction of molecular interactions but also accelerate the identification of viable candidates for further evaluation.
Complementary strategies, including combinatorial chemistry and high-throughput screening (HTS), enable the rapid generation and assessment of large chemical libraries, further shortening timelines for identifying promising leads [5]. Natural products continue to provide a rich reservoir of structurally unique compounds, with microorganisms, plants, and marine organisms contributing to the discovery of novel bioactive agents [6,7]. Collectively, these approaches illustrate how modern drug design blends traditional sources with computational advances to expand therapeutic options. Moreover, ADMET profiling ensures that candidate molecules are not only effective but also safe, thereby increasing the likelihood of clinical success [8].
By integrating historical examples such as statins with contemporary approaches like CADD and AI-driven methods, this review aims to provide a comprehensive overview of the strategies, applications, and challenges that define drug design in the current era.
AIM AND OBJECTIVE
The primary aim of this review is to provide an integrated understanding of drug design and discovery, highlighting both theoretical concepts and practical applications. An important objective is to emphasize chemical management and safety guidelines, including appropriate storage, labeling, disposal, and emergency response strategies, which are essential for minimizing risks in laboratory environments. In addition, the review seeks to enhance knowledge of reaction preparation and purification methods, focusing on techniques such as distillation, crystallization, chromatography, and filtration, which ensure the acquisition of pure and well-characterized compounds.
Another key objective is to outline the major phases of drug discovery, including target identification and validation, hit identification, lead optimization, and preclinical and clinical development, while underscoring the collaborative and multidisciplinary nature of the process [1]. Particular attention is given to physicochemical properties and their role in Quantitative Structure– Activity Relationship (QSAR) studies, covering the use of molecular descriptors, statistical modeling techniques, and model validation [2,19]. Furthermore, the review examines different methods of structure-based drug design, such as molecular docking and de novo design, highlighting their principles, applications, and limitations in identifying and refining potential drug candidates [3,4].
The objectives also extend to the study of natural products as a continuing source of therapeutic agents, including their extraction, isolation, structural determination, and evaluation of pharmacological activity [6,7]. By combining insights from chemical management, reaction methodology, drug discovery phases, QSAR, structure-based design, and natural product exploration, this review aims to present a unified perspective of pharmaceutical development. Such integration not only reinforces theoretical knowledge but also develops practical competencies that are essential for advancing drug discovery research.
DRUG DESIGN AND DEVELOPMENT -
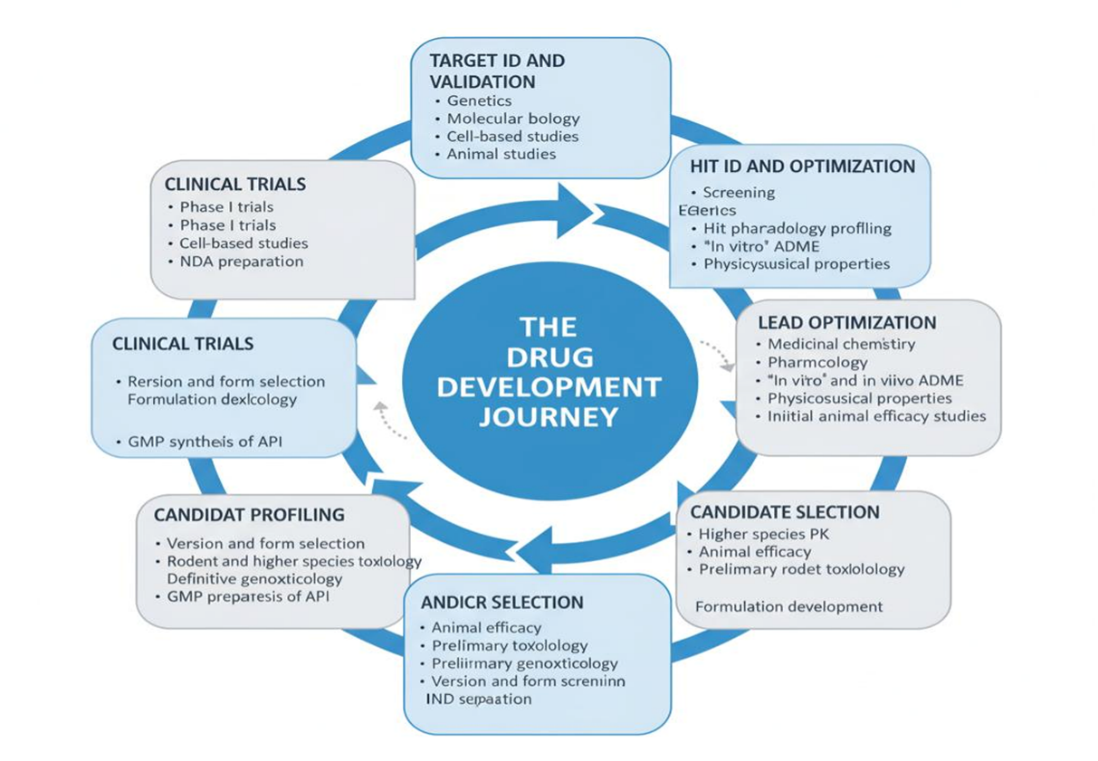
Drug discovery and development is a complex, multi-stage process that integrates chemistry, biology, pharmacology, and computational sciences to transform an initial idea into a therapeutic agent. The stages include target identification, hit discovery, lead optimization, preclinical evaluation, regulatory submission, and clinical trials. Each step is crucial, as attrition rates in drug discovery are high, and only a fraction of compounds ever reach the market [1].
The first stage in drug discovery involves identifying a biological target, such as an enzyme, receptor, or protein implicated in disease pathology. Advances in genomics, proteomics, and bioinformatics have accelerated this process, enabling researchers to identify and validate disease associated targets more effectively [1,5]. Target validation ensures that modulation of the selected biomolecule will have a therapeutic effect without causing unacceptable side effects. Natural products and secondary metabolites have historically contributed significantly to drug discovery, and even in the genomic era, they remain valuable as novel sources of target modulators [6,7].
Once a validated target is identified, the next step is to discover molecules, known as hits, that can interact with it. High-throughput screening (HTS) enables the rapid evaluation of thousands to millions of compounds against a target to identify initial hits [5]. In parallel, virtual screening techniques employ computational methods to filter large chemical libraries for compounds predicted to bind effectively [4]. Increasingly, AI-based algorithms are also being employed to enhance hit identification by predicting molecular interactions and prioritizing promising compounds [10].
Hit compounds are subsequently refined into lead molecules with improved potency, selectivity, and drug-like properties. Medicinal chemists employ Quantitative Structure–Activity Relationship (QSAR) models, which establish correlations between chemical structures and biological activity, allowing systematic modification of functional groups to enhance activity [2]. Structure-based drug design uses crystallographic or computational models of the target protein to optimize binding interactions [4]. De novo drug design approaches also contribute by generating novel compounds from scratch, tailored to the target binding site [3,9]. AI and machine learning approaches are now automating parts of this process, accelerating the design of new scaffolds and optimizing pharmacokinetic properties [10,11].
Modern drug design increasingly depends on computational techniques to simulate and predict drug–target interactions. Molecular docking provides insights into the preferred orientation and binding affinity of small molecules within a target protein’s active site [4]. QSAR models mathematically correlate chemical structures with biological activities, enabling activity prediction for novel compounds [2]. De novo drug design tools construct novel molecular entities from scratch, guided by target site characteristics [3,9]. These approaches, combined with AI-driven methods such as explainable artificial intelligence and large language models, are transforming drug discovery by improving hit-to-lead predictions and suggesting innovative chemical scaffolds [10,11].
After promising lead compounds are identified, preclinical studies are conducted to evaluate pharmacological potential and eliminate molecules with poor efficacy or safety. These studies involve a combination of in vitro assays using cell and tissue cultures, biochemical experiments such as isothermal titration calorimetry to study protein–ligand interactions, and in vivo animal studies using models with close physiological resemblance to humans [1,5]. In addition to experimental methods, ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) modeling is increasingly applied to predict pharmacokinetic and safety profiles at early stages, thereby reducing the risk of clinical failure [18]. The goal of preclinical testing is to generate a robust safety and efficacy profile that supports progression to human trials.
If preclinical studies demonstrate favorable safety and pharmacological results, an Investigational New Drug (IND) application is submitted to the U.S. Food and Drug Administration (FDA) or other regulatory bodies. This application contains comprehensive preclinical data, including pharmacology, toxicology, manufacturing, and quality control information, to justify the initiation of human clinical trials [1]. The sponsor organization, which may be a pharmaceutical company, academic institution, or collaborative group, is responsible for preparing and submitting the IND. Only after regulatory approval of the IND can the investigational compound move forward into clinical trials.
Clinical trials are prospective studies involving human volunteers or human-derived specimens that are conducted to answer specific research questions about a drug candidate. These studies assess the safety, efficacy, and therapeutic effectiveness of new drugs or biological interventions [1]. Clinical trials proceed through multiple phases:
Phase I focuses on safety, tolerability, and pharmacokinetics in a small group of healthy volunteers. Phase II evaluates efficacy and side effects in a larger patient population.
Phase III confirms therapeutic benefit and compares the investigational drug against existing standard treatments in diverse patient groups.
Upon successful completion of clinical trials, regulatory authorities review the data and, if satisfactory, approve the drug for clinical use.
Approaches to drug design
[A] QSAR: Quantitative Structure-Activity Relationship
Definition
Quantitative Structure-Activity Relationship (QSAR) is a computational modeling technique that employs mathematical and statistical approaches to predict the biological activity, physicochemical properties, or environmental behavior of chemical compounds based on their molecular structure [2,19]. The central principle of QSAR is that quantifiable molecular descriptors can be correlated with experimentally observed biological or chemical activities. By analyzing these correlations, QSAR models assist researchers in designing and optimizing new compounds with desired characteristics, thereby accelerating the drug discovery and development process [1,9,19].
Key Parameters in QSAR
Hydrophobicity (Log P): Hydrophobicity is expressed as the logarithm of the partition coefficient (Log P) between water and a nonpolar solvent such as octanol. It reflects the compound’s preference for lipid versus aqueous environments. Molecules with balanced hydrophobicity typically show improved absorption and distribution, making Log P a critical parameter in evaluating drug-likeness [2,18,20].
Electronic Effects (Hammett Constant): The Hammett constant (σ) measures the electron-donating or electron-withdrawing influence of substituents on a benzene ring. These effects influence molecular reactivity and interactions with biological targets, impacting biological activity [2,20].
Steric Effects (Taft’s Steric Factor): Taft’s steric factor (Es) quantifies spatial hindrance caused by substituents near reactive sites. Steric effects determine how well a molecule fits into the active site of a target enzyme or receptor, affecting binding affinity and biological activity [2,20].
Molecular Weight: Molecular weight impacts pharmacokinetic properties including absorption, distribution, metabolism, and excretion (ADME). Compounds with moderate molecular weight (300–500 Daltons) generally exhibit favorable ADME profiles [18].
Dipole Moment: The dipole moment measures charge separation in a molecule, reflecting polarity. Compounds with optimal polarity interact effectively with biological targets and aqueous environments, influencing solubility and binding efficiency [18].
Number of Aromatic Moieties: Aromatic rings contribute to the stability and specificity of drugtarget interactions. The number and type of aromatic moieties can affect binding affinity, selectivity, and overall biological activity [2,20].
Process and Examples
Examples of QSAR Applications:
Drug Design: QSAR models guide drug design, such as optimizing chloroquine analogues for antimalarial activity, enhancing efficacy while reducing resistance [1,2,19].
Toxicity Prediction: QSAR predicts toxic effects of chemicals and environmental pollutants, for example, LC50 values of pesticides [18,21].
Environmental Chemistry: QSAR predicts chemical behavior in the environment, including biodegradability and bioaccumulation, supporting regulatory decisions [18,21].
Tools Required for QSAR
QSAR Toolbox: Developed by OECD, it supports chemical hazard assessment, experimental data retrieval, metabolism simulation, and profiling chemical properties, facilitating QSAR model development and validation [18,19].
Chemical Databases: PubChem, ChemSpider, and ChEMBL provide essential molecular descriptors and experimental data for QSAR analysis [18].
Computational Chemistry Software: Programs like Gaussian, Spartan, and AutoDock calculate molecular descriptors and perform molecular docking simulations, improving QSAR predictive accuracy [9,18,19].
Types of QSAR
Advantages of QSAR
Efficiency: QSAR accelerates drug discovery by predicting compound activity without extensive laboratory testing [1,9,19].
Cost-Effectiveness: Reduces reliance on expensive and time-consuming experiments [18,19].
Predictive Capability: Provides insights into structure-activity relationships, guiding the design of safer and more effective compounds [2,10,19].
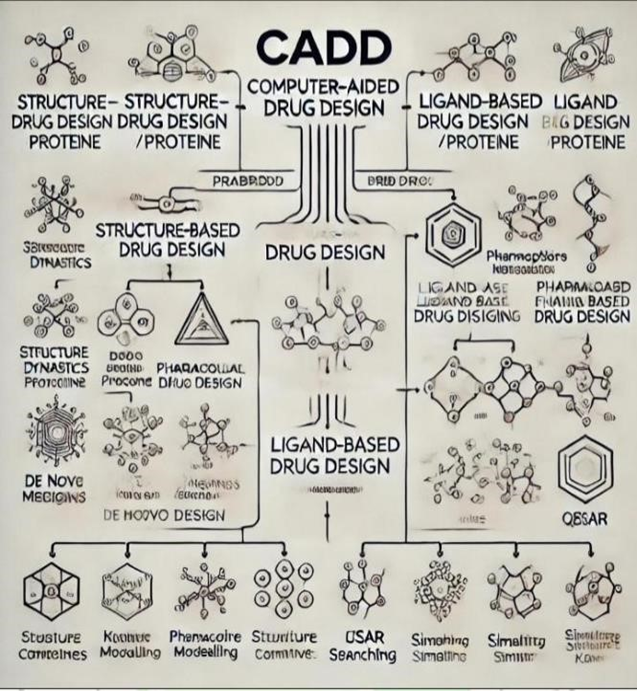
[B] ( Computer-Aided Drug Design )
Computer-Aided Drug Design (CADD) is an advanced computational approach used to facilitate drug discovery and development. By employing algorithms and computational tools, CADD predicts how drug molecules interact with biological targets, thereby accelerating the identification of potential drug candidates while reducing experimental costs and efforts (1,3,4). It plays a pivotal role in modern drug development by enabling in silico simulation and analysis of molecular interactions, structure-activity relationships (SAR), and drug efficacy before experimental testing (2,19). A widely used application of CADD is Structure-Based.
Drug Design (SBDD), which utilizes the three-dimensional structure of biological targets, such as enzymes or receptors, to design molecules that can effectively bind to them (4). For example, inhibitors targeting HIV protease or cancerrelated kinases often rely on SBDD (4,5). Another approach, Ligand-Based Drug Design (LBDD), uses information from known active compounds to design new molecules with similar biological activity (3,19). Common techniques in CADD include molecular docking, virtual screening, pharmacophore modeling, and QSAR analysis (2,19).
Key CADD-related terms include molecular modeling (visualizing and simulating molecular structures), docking (predicting optimal drug-target binding poses), and pharmacokinetics (studying drug absorption, distribution, metabolism, and excretion) (8). Notable drugs developed using CADD include imatinib (Gleevec) for chronic myeloid leukemia and oseltamivir (Tamiflu) for influenza (6,7). By leveraging computational power and sophisticated algorithms, CADD has transformed drug discovery, allowing for more efficient and accurate drug design while minimizing trial-and-error experiments (3,9,10).
Structure-Based Drug Design (SBDD): Utilizes the 3D structure of a target protein to design drugs that bind effectively to its active site (4).
Ligand-Based Drug Design (LBDD): Designs or identifies new molecules based on the knowledge of ligands that interact with the target, focusing on patterns in molecular activity (3).
Chemical Structure Drawing in CADD
Chemical structure drawing is a fundamental part of CADD, allowing researchers to visually represent chemical compounds and understand their properties and interactions with biological targets (3,4).
Typical steps involved:
Key Tools: ChemSketch (1), PyMOL (3), UCSF Chimera (3), Jmol (3), and Marvin Sketch (1).
Computational Drawing in CADD
Computational drawing involves creating detailed 2D and 3D models of molecules and biological targets using specialized software. This facilitates visualization of molecular interactions, optimization of drug candidates, and prediction of their behavior in biological systems (3,4,9).
Key Tools:
Chemical Structure Presentation
Chemical structure presentation is crucial for accurately communicating molecular information in research and education (3).
The process involves:
1. Initial Sketching: Drawing the basic molecular skeleton using tools like ChemSketch and MarvinSketch (1).
2. Functional Group Addition: Adding specific functional groups to define chemical properties (1).
3D Visualization: Using PyMOL, UCSF Chimera, or Jmol to study spatial arrangement and interactions (3,4).
3. Property Calculation: Computing molecular properties such as weight, hydrophobicity (Log P), and hydrogen bond donors/acceptors to predict biological activity (20,21)
4. Structure Optimization: Ensuring molecules adopt stable, low-energy conformations (3).
5. Reaction Drawing: Illustrating reactants, products, and reaction conditions for synthetic pathways (1).
6. File Formats and Export: Saving structures in formats like .mol, .sdf, or .pdb for compatibility with computational tools (3).
Popular Tools: ChemSketch (1), PyMOL (3), UCSF Chimera (3), Jmol (3), MarvinSketch (1), ChemDraw (1), BIOVIA Draw (3), MOE (3), Avogadro (3), ChemDoodle (1).

Chemical Databases
Chemical databases are vital resources for researchers, chemists, and pharmaceutical scientists, providing comprehensive information on chemical compounds, their properties, and biological activities (5,6). The internet has greatly expanded access to these resources, and new databases continue to emerge alongside established ones, offering innovative tools for drug discovery and chemical research (5,7).
Some of the widely used chemical databases include:
These databases support the design, optimization, and analysis of potential therapeutic molecules by providing reliable, searchable, and up-to-date chemical information (3,5,7).
[C] Pharmacophore models
Computer-Aided Drug Discovery (CADD) uses computational methods and data resources to investigate molecular properties and develop new therapeutic agents. Broadly, it involves designing or selecting compounds as potential drug candidates before synthesis and experimental testing, helping reduce costs and research time ([3],[4],[10]). Traditional in vitro screening is expensive and time-consuming, making computational alternatives highly valuable. Virtual Screening (VS) is a CADD technique that screens chemical libraries in silico to identify compounds most likely to interact with a target, which can be accelerated using pharmacophore models as queries to select molecules with desired features ([3],[4]).
Pharmacophore modeling is based on the principle that compounds with common chemical functionalities arranged similarly in space often exhibit activity against the same biological target. These features are represented as geometric entities—such as spheres, planes, or vectors—in the pharmacophore model. Key features include hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), hydrophobic regions (H), positively and negatively ionizable groups (PI/NI), aromatic rings (AR), and metal-coordinating regions ([4],[23]). Shape or exclusion volumes (XVOL) can be added to define steric constraints of the binding pocket.
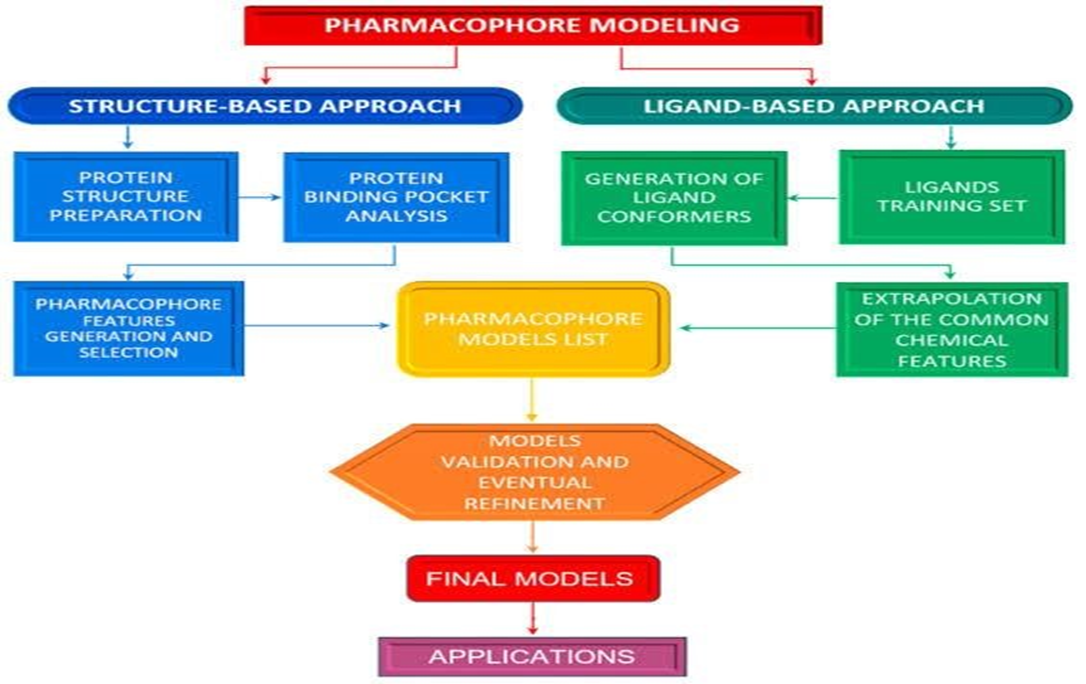
Structure-Based Pharmacophore Modeling: This method relies on the 3D structure of a protein target. Atomic-level details extracted from the protein’s holo or apo form allow the identification of stereo-electronic features responsible for ligand activity ([4],[23]).
Ligand-Based Pharmacophore Modeling: This approach starts with active compounds that bind the same target. Since the bioactive conformations of these ligands may be unknown, multiple conformers are generated to ensure at least one represents the biologically active form. Shared chemical features from these conformers are then used to construct the pharmacophore ([4],[23]).
Molecular docking predicts how a ligand binds to a protein target. A 3D structure of the protein, often determined by X-ray crystallography or NMR spectroscopy, is required ([3],[4]). The protein and a library of ligands are used as input for docking programs.
Docking depends on two main components: the search algorithm, which explores possible ligand orientations and conformations relative to the protein, and the scoring function, which estimates binding affinity. Exhaustively searching all conformations and orientations is computationally impossible. Most programs account for flexible ligands, and some also incorporate flexible protein receptors to improve prediction accuracy ([3],[23]).
Recent docking innovations include AI-assisted methods and flexible receptor modeling to increase accuracy and speed in virtual screening ([23],[24]).
ADMET properties—absorption, distribution, metabolism, excretion, and toxicity—are critical in assessing drug candidates. An effective compound must not only target the intended receptor but also demonstrate suitable ADMET characteristics at therapeutic doses ([8],[23]).

Numerous in silico models predict ADMET properties. One approach is the ADMET-score, which evaluates drug-likeness based on 18 ADMET endpoints predicted via the admetSAR web server. Each property is weighted according to model accuracy, pharmacokinetic importance, and usefulness ([23],[24]).
Performance was evaluated using FDA-approved drugs, ChEMBL small molecules, and drugs withdrawn due to safety concerns. Analysis showed significant differences among these datasets, with no clear linear correlation between the ADMET-score and quantitative estimate of druglikeness (QED) ([23]).
For scoring, beneficial endpoints (e.g., hERG–) are assigned a value of 1, while harmful endpoints (e.g., hERG+, Ames, CARC, CYP inhibitors, OCT2, P-gp inhibitors) are assigned 0. The ADMETscore is normalized between 0 and 1, where 1 represents the most favorable profile. Scores below 0 are set to 0, and those above 1 are capped at 1 ([23],[24]). This system allows prioritization of compounds with optimal pharmacokinetic and safety profiles.
[D] Combinatorial Chemistry
Combinatorial chemistry is a strategy that generates a large collection of structurally diverse compounds, known as a chemical library, by systematically linking different “building blocks” through repetitive covalent reactions [25]. Once synthesized, these compounds can be simultaneously screened for their interactions with specific biological targets [25,26]. Active compounds can be identified directly in position-addressable libraries or indirectly through decoding techniques using chemical or genetic methods. Common approaches include phage display, yeast display, bacterial display, mRNA display, one-bead-one-compound (OBOC), DNAencoded chemical libraries (DECL), and solution-phase mixture libraries [25,28].
Besides creating a vast number of compounds, these combinatorial methods allow rapid concurrent screening against specific drug targets [25,29]. Lower-throughput techniques, such as parallel synthesis libraries and planar microarray libraries, produce more focused libraries [25]. Planar microarrays are mainly used for peptide research but, in principle, can be adapted for other compound types through automation. Focused small-molecule libraries, typically consisting of hundreds to thousands of compounds, are especially valuable for lead optimization when combined with computational chemistry [25,26,30].
High-Throughput Screening (HTS) involves testing a large number of potential bioactive compounds against a target biological process or event [5,25]. HTS is applied to combinatorial chemistry libraries, including genomic, protein, peptide, and DNA-encoded libraries (DECL) [5,25,28]. The main aim is to accelerate drug discovery by screening vast numbers of compounds, often hundreds of thousands, at rates exceeding 20,000 compounds per week [5,25]. Advanced HTS relies on assay adaptation, robotic systems, and optimized implementation strategies. UltraHigh-Throughput Screening (UHTS) extends this further, enabling the testing of up to 100,000 compounds per day while reducing costs, manipulation steps, and enhancing efficiency [25,29,30].
Combinatorial chemistry combined with HTS has become a key method for identifying active compounds from large chemical libraries [25,26]. Integration with Quantitative Structure-Activity Relationship (QSAR) studies helps reduce drug development costs by screening compounds virtually, filtering out those predicted to be toxic or having poor pharmacokinetic profiles [2,19,25,27]. Techniques include classical linear methods, such as partial least squares, as well as nonlinear approaches [2,19]. Increasingly, machine learning and pattern recognition methods are being applied to rapidly and accurately assess large compound datasets, supported by chemoinformatics tools for QSAR modeling [10,26,30].
Drug discovery involves identifying and developing new therapeutic agents from diverse sources [1,6,7]:
1. Natural Products:
Plants, microorganisms, animals, and marine organisms have historically provided bioactive compounds [6,7]. Plants yield secondary metabolites such as alkaloids, flavonoids, and glycosides—for example, morphine from Papaver somniferum and paclitaxel from Taxus brevifolia [6]. Microorganisms produce antibiotics like penicillin from Penicillium notatum and streptomycin from Streptomyces griseus [6]. Marine organisms have emerged as a source of novel drugs, such as ziconotide from cone snail venom and trabectedin from sea squirts [7]. Animals have also inspired drugs, like captopril, derived from the venom of the Brazilian pit viper [6,7].
Synthetic approaches allow the creation of entirely new molecules or modifications of natural compounds to improve their properties [1]. Aspirin, for instance, is a synthesized derivative of salicylic acid from willow bark [1]. Synthetic drugs such as propranolol (beta-blocker) and fentanyl (opioid) illustrate the scalability and versatility of this approach [1]
Genetic engineering enables the production of biologics, including recombinant proteins and monoclonal antibodies [1,10]. Examples include insulin and adalimumab, which are effective against complex conditions like cancer and autoimmune diseases [1,10].
Applications of Drug Design
Drug design, whether structure-based, ligand-based, or computationally aided, is a cornerstone of modern pharmaceutical research. Its applications span the discovery, optimization, and development of therapeutic agents, improving efficiency, reducing costs, and minimizing trialand-error in drug development [31,32,33].
Drug design enables the rational creation of molecules that selectively interact with biological targets, including enzymes, receptors, or nucleic acids. Structure-based drug design (SBDD) has been instrumental in developing inhibitors for HIV protease, tyrosine kinases, and other cancerrelated targets [33,34]. Ligand-based drug design (LBDD) assists in developing compounds when the 3D structure of the target is unknown by utilizing information from known active ligands [33,34]. Computational tools such as molecular docking and pharmacophore modeling help predict binding affinities and interactions, accelerating the identification of potent candidates [34,35]. Examples: Imatinib (Gleevec) for chronic myeloid leukemia and Oseltamivir (Tamiflu) for influenza were developed using CADD approaches [31,33].
Combinatorial chemistry combined with HTS allows rapid evaluation of large compound libraries. Structurally diverse molecules are generated systematically, enabling thousands to millions of compounds to be screened efficiently [25,26,28,30]. DNA-encoded chemical libraries (DECL) and one-bead-one-compound (OBOC) libraries are commonly used for hit identification [28,30]. Computational filtering and virtual screening help prioritize compounds with favorable pharmacokinetic and toxicity profiles, reducing experimental screening costs and time [27,31].
After hit identification, drug design facilitates lead optimization to improve efficacy, selectivity, and safety. Molecular dynamics simulations, QSAR modeling, and ADMET predictions guide structural refinement [2,19,23,26,27]. Predictive tools, such as ADMET scoring and AI-based models, allow simultaneous evaluation of multiple candidates, enhancing the selection of promising leads [23,26,27,31].
Drug design aids in identifying and optimizing bioactive compounds from natural sources such as plants, microorganisms, marine organisms, and animals [6,7,31]. Rational modifications improve efficacy, bioavailability, and reduce toxicity.
Examples: Morphine from Papaver somniferum and paclitaxel from Taxus brevifolia [6] Antibiotics like penicillin from Penicillium notatum and streptomycin from Streptomyces griseus.[6] Marine-derived drugs such as ziconotide from cone snail venom [7]
Synthetic chemistry and biotechnology benefit from drug design by enabling the creation of new molecules or modifying existing compounds [31,32].
Examples: Aspirin, a synthetic derivative of salicylic acid, designed for improved efficacy [31] Beta-blockers like propranolol and opioids like fentanyl illustrate scalability [31] Biologics, including recombinant insulin and monoclonal antibodies (e.g., adalimumab), optimized using computational modeling and AI [10,26,31]
In silico ADMET modeling predicts absorption, distribution, metabolism, excretion, and toxicity, prioritizing drug-like compounds while eliminating those with adverse effects [23,26,27,31]. Tools like ADMETlab 3.0 and AI-based models enable rapid, large-scale evaluations, saving time and cost in early drug development [23,26,27,31].
CADD and AI-driven approaches facilitate personalized therapies by predicting patient-specific responses based on genetic, metabolic, and proteomic data. This enhances efficacy and minimizes adverse effects, contributing to precision medicine [10,11,31].
“While these approaches and applications highlight the potential of modern drug discovery, it is equally important to recognize the challenges and limitations that continue to constrain progress.”
CHALLENGES AND LIMITATIONS
Despite significant advances in technology and methodology, drug discovery continues to face multiple challenges and limitations that hinder efficiency and innovation. One of the most persistent obstacles is the high cost and extended development timelines. It is estimated that bringing a single drug to market may take more than a decade and require billions of dollars in investment [35]. Although new technologies such as automation and AI are increasingly adopted, the paradox remains that drug development has not become substantially more cost-effective or time-efficient [35].
Another major limitation is the high attrition rate in clinical trials, where the majority of drug candidates fail to progress to approval. Failures often occur due to unforeseen safety concerns, poor efficacy, or suboptimal pharmacokinetic properties, reflecting the unpredictable nature of complex biological systems [34]. This high failure rate underscores the inherent risks of drug discovery and the urgent need for more predictive preclinical models.
The integration of artificial intelligence (AI), while promising, also presents its own set of challenges. Issues related to data quality, bias, model interpretability, and ethical considerations continue to restrict the widespread adoption of AI in drug development [36]. Furthermore, the complexity of biological networks often exceeds the current capabilities of computational models, limiting their predictive accuracy [36].
Regulatory hurdles add another layer of difficulty, as evolving policies and the absence of clear guidance for novel therapeutics frequently delay approvals [37]. These delays not only extend development timelines but also impact patient access to innovative therapies. Similarly, polypharmacology—designing drugs that act on multiple targets—offers therapeutic potential but raises concerns about unintended off-target effects, which can increase the risk of adverse outcomes [38].
Equally pressing are the challenges in drug delivery systems, where ensuring stability, targeted release, and biological compatibility remain major barriers. Despite progress, scalability and reproducibility continue to impede the successful translation of novel delivery platforms into clinical practice [39]. These issues are compounded by the complexity of disease mechanisms themselves, as incomplete understanding of disease biology complicates target identification and drug design [40].
Finally, academic drug discovery faces systemic limitations. Many universities and research institutes struggle with inadequate funding, limited infrastructure, and lack of prioritization for translational drug development [41]. As a result, promising discoveries in the academic setting often fail to progress toward commercialization, highlighting the gap between early-stage research and industrial application.
Collectively, these challenges—ranging from economic constraints and scientific uncertainty to regulatory and infrastructural barriers—underscore the complexity of drug discovery. Addressing them will require interdisciplinary collaboration, integration of advanced technologies, and adaptive regulatory frameworks to accelerate the translation of innovative ideas into safe and effective therapies.
CONCLUSION
Drug design and discovery remain a dynamic and rapidly advancing discipline that is central to improving healthcare and patient outcomes [1,3,6]. Traditional strategies, such as the use of natural products, continue to play an important role, but they are now complemented by modern innovations including artificial intelligence, genomics, and nanotechnology, which are reshaping the way novel therapeutics are conceived and developed [7,9–11]. Each phase of the discovery pipeline—from target identification and validation through to preclinical and clinical evaluation— illustrates the synergy between scientific insight and technological advancement [1,3,4].
Nevertheless, persistent barriers such as high costs, long development timelines, and high attrition rates remain major limitations to efficiency [34,35]. The growing threat of drug resistance further underscores the urgency for new discovery strategies. Promising solutions are emerging through the application of machine learning, precision medicine, sustainable “green chemistry” approaches, and advanced drug delivery systems [10,11,26,39]. Together, these innovations are enabling more predictive, efficient, and safer drug development pathways.
Ultimately, drug discovery exemplifies human ingenuity in addressing the complexities of disease biology. Its interdisciplinary nature—spanning chemistry, biology, data science, and clinical medicine—ensures a continuous flow of innovation with the potential to reshape medical treatment and significantly improve global health outcomes [1,3,5,11].
REFERENCES


Pragati Nade, Rohini Satdive, Prachi Kadam, Renuka Sagane, Sabafarin Shaikh, Rational Drug Design and Development: Trends and Future Directions, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 11, 2288-2309. https://doi.org/10.5281/zenodo.17615793
 10.5281/zenodo.17615793
10.5281/zenodo.17615793
