Department of Pharmaceutical Chemistry, College of Pharmacy, Madras Medical College, Chennai.
The Janus kinase 3 (JAK3) pathway is essential for immunological control and inflammatory reactions, it is a viable therapeutic target for inflammatory bowel disorders (IBD), such as Crohn's disease (CD) and ulcerative colitis (UC). As possible JAK3 inhibitors, this study investigates in silico methods for designing and assessing new quinazoline derivatives. The pharmacological actions of quinazoline scaffolds are well-known, and their modification presents chances for JAK3 inhibition that is selective. After a library of quinazoline derivatives was created using computational drug design approaches, they were molecularly docked to the JAK3 crystal structure (PDB ID:5toz). The best binding properties of the derivatives were found by analyzing pharmacophore alignment, interaction patterns, and binding affinities. Additional insilico evaluations included ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiling to forecast drug-like characteristics and molecular dynamics simulations to assess the stability of ligand-receptor complexes. High specificity for JAK3 and acceptable pharmacokinetic profiles were displayed by a few chosen derivatives, reducing the possibility of off-target effects.
Ulcerative colitis (UC) was initially identified as one of the two main types of inflammatory bowel disease (IBD). Mucosal inflammation in UC is characterized by a continuous extension of inflammation from the rectum proximally in the colon. The other form of IBD, Crohn's disease (CD), exhibits patchy lesions that may be dispersed throughout the gastrointestinal tract. While CD is characterized by transmural inflammation (including all layers of the intestinal wall) that results in fibrosis, stricture, and fistula, UC is usually limited to inflammation of the mucosal layer, causing superficial damage of the gut wall [1].
The image illustrates these conditions by showing inflamed areas in the intestines, helping to differentiate the affected regions and patterns of inflammation in both Crohn's disease and Ulcerative Colitis.
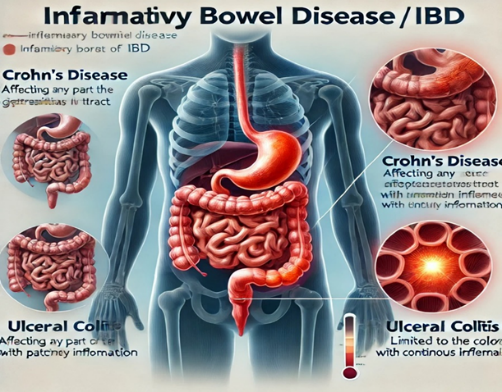
Figure 1: Inflammation in UC and CD
Signs and symptoms of ulcerative colitis [2]:
People with ulcerative colitis typically experience diarrhoea and abdominal pain. They may also have additional symptoms, such as:
Some people may also show signs of a low red blood cell count (anemia) due to blood loss.
Symptoms of crohn’s diseases:
Treatment for ulcerative colitis and crohn’s diseases [3]
IBD cannot be cured, although there are a number of treatments that can help control its symptoms:
Medications: Biologics, immune system suppressors, and anti-inflammatory medications are frequently used.
Surgery: Surgical procedures may be necessary for certain patients.
Selection of target
Why JAK3 in Particular?
Selectivity for ?c Cytokines: JAK3 is more focused than JAK1 or JAK2 inhibitors, which may impact other pathways and enhance side effects, because it is mostly linked to ?c receptors.
Effects Specific to Lymphocytes: JAK3 targeting directly affects immune cell function, which is essential for reducing inflammation in IBD.
mechanism of action of JAK3[4]:
IBD overactivation:
Barrier dysfunction:
JAK3 inhibitors in ulcerative colitis and crohn’s diseases: mechanism, role and therapeutic potential
Role of JAK inhibitors in UC and CD [5]:
mechanism of action of JAK3 inhibitors:
JAK3 Inhibition [6]
Signal Transducer and Activator of Transcription (STAT) proteins cannot be phosphorylated or activated when JAK3 inhibitors are present because they interfere with JAK3 action.
Modulation of Immune Cell Function:
Pharmacophore modeling [7]
Pharmacophore modeling studies have become one of the tools in the field of drug discovery. In 1909, Paul Ehrlich introduced the concept of Pharmacophore, who defined the pharmacophore as a molecular framework that carries the essential features responsible for a drug’s biological activity. The IUPAC defines “A pharmacophore is ensemble of stearic and electronic features that is necessary to ensure the optimal supra-molecular interaction with a specific biological target and to trigger or block its biological response”.
Pharmacophore features
Selection of scaffold
Heterocyclic ring system: Heterocyclic systems are recognized to be of great importance due to their proven utility within the field of medicinal chemistry. Heterocyclic compounds are cyclic rings of atoms that contain at least one heteroatom. The most frequent hetero-atoms are nitrogen, oxygen, and Sulphur, but heterocyclic rings including additional hetero-atoms i.e., phosphorus, iron, magnesium, selenium, etc. are also common. Heterocycles are the most important traditional division of organic chemistry, and research interest in heterocycles is increasing because of their medicinal applications [8].
Nitrogen-based heterocyclic chemistry has recently piqued the interest of medicinal chemists looking forward to the design of novel and effective medicinal compounds. Various nitrogen-based heterocycles such as pyrroles, pyrazoles, imidazoles, triazoles, pyridine, pyrazine, pyrimidine, etc. have been reported in the scientific databases possessing multiple physiological and pharmacological properties.
Selection of quinozoline as scaffold:
Study Objective
Designing and evaluating novel quinazoline derivatives as potential JAK3 inhibitors for UC and CD.
MATERIALS AND METHOD:
The ligands were drawn using the ChemSketch software based on the necessary pharmacophoric features. Zinc15 and the Pubchem database were used to verify the novelty of the compounds. The designed compounds were considered to be novel since there is no data available in the ZINC® database.
2.selection of biological target:
A Protein Data Bank is a crystallographic database for three-dimensional structural data of large biological molecules such as Protein, Nucleic acid and Complex assemblies. In this study, janus kinase 3 inhibitors were Chosen as a target for the treatment of UC and CD. The efficient PDB enzyme target was selected with lower resolution (PDB ID: 5toz) provides a significant target for UC and CD.
Preparation of protein:
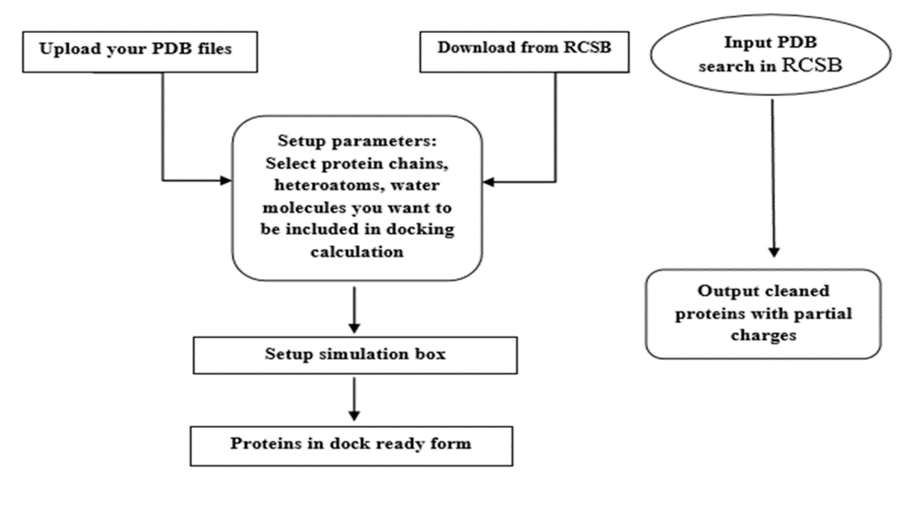
Figure 2: Preparation of protein
3. In-silico screening of drug likeness property:
Swiss ADME is a free web tool that predicts the Absorption, Distribution, Metabolism, and Excretion (ADME) properties of small molecules, along with other pharmacokinetic and physicochemical characteristics. It is widely used in drug discovery and development to evaluate a compound’s drug-likeness and optimize its pharmacological properties.
4. In-silico screening of toxicity prediction:
The Osiris Property Explorer is a computational tool designed to predict the drug-relevant properties of chemical compounds. It provides a quick and intuitive way to estimate various molecular properties, including toxicity risks, physicochemical properties, and drug-likeness. It is widely used in the early stages of drug discovery and development for compound optimization.
5. Docking protocol:
The interaction between a tiny molecule (ligand) and a target macromolecule (usually a protein or nucleic acid) can be predicted computationally using a technique called molecular docking. By simulating their binding posture and affinity, it is a vital technique in structure-based drug design that helps find possible drug candidates.
Post docking evaluation:
Molegro Molecular Viewer (MMV) is an easy-to-use software program for examining protein-ligand interactions and viewing molecular structures. It provides a range of visualization options to examine binding poses, interaction networks (hydrophobic, electrostatic, and hydrogen bonding), and molecular characteristics. It is especially useful for post-docking research [10].
Synthetic scheme:
Scheme 1: for compounds (1-22)
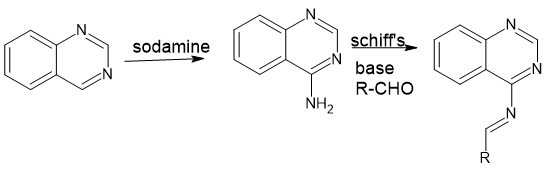
Scheme 2: for compounds (23-51)
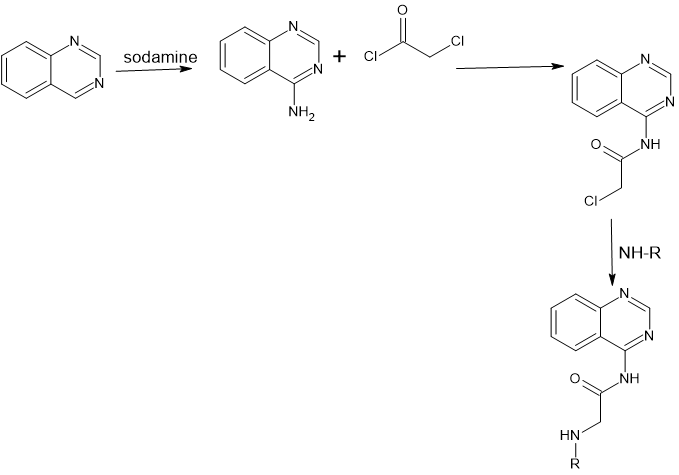
Novelty assessment:
The designed 51 compounds were considered to be novel since there is no data available in the ZINC® and the Pubchem database.
In-silico screening of drug-likeness property:
As per Lipinski rule of 5, ADME properties of novel proposed analogues were assessed using Swiss ADME, a free web tool.
Table No.1: Drug-likeness property of novel proposed analogues
|
Compound code |
Log P |
Molecular weight |
No. of HBA |
No. of HBD |
No. of Rot. bonds |
Lipinski rule of 5 (n violation) |
|
PD1 |
2.50 |
233.27 |
3 |
0 |
2 |
0 |
|
PD2 |
2.80 |
263.29 |
4 |
0 |
3 |
0 |
|
PD3 |
2.72 |
247.79 |
3 |
0 |
2 |
0 |
|
PD4 |
2.76 |
281.74 |
3 |
0 |
2 |
0 |
|
PD5 |
2.66 |
261.32 |
3 |
0 |
4 |
0 |
|
PD6 |
2.54 |
217.22 |
5 |
0 |
3 |
0 |
|
PD7 |
2.00 |
223.24 |
4 |
0 |
2 |
0 |
|
PD8 |
2.19 |
249.27 |
4 |
1 |
2 |
0 |
|
PD9 |
2.76 |
247.27 |
3 |
0 |
2 |
0 |
|
PD10 |
2.08 |
278.27 |
5 |
0 |
3 |
0 |
|
PD11 |
2.90 |
293.32 |
5 |
0 |
4 |
0 |
|
PD12 |
2.99 |
277.32 |
4 |
0 |
3 |
0 |
|
PD13 |
2.75 |
301.27 |
6 |
0 |
3 |
0 |
|
PD14 |
2.16 |
278.27 |
5 |
0 |
3 |
0 |
|
PD15 |
2.71 |
297.74 |
4 |
1 |
2 |
0 |
|
PD16 |
2.17 |
283.71 |
4 |
1 |
2 |
0 |
|
PD17 |
2.70 |
318.19 |
3 |
0 |
2 |
0 |
|
PD18 |
2.77 |
267.71 |
3 |
0 |
2 |
0 |
|
PD19 |
2.41 |
239.30 |
3 |
0 |
2 |
0 |
|
PD20 |
2.11 |
278.27 |
5 |
0 |
3 |
0 |
|
PD21 |
3.28 |
289.37 |
3 |
0 |
5 |
0 |
|
PD22 |
2.20 |
261.28 |
4 |
0 |
3 |
0 |
|
PD23 |
1.70 |
278.31 |
3 |
2 |
5 |
0 |
|
PD24 |
1.85 |
348.19 |
4 |
2 |
5 |
0 |
|
PD25 |
1.30 |
279.30 |
4 |
2 |
5 |
0 |
|
PD26 |
1.90 |
308.33 |
4 |
2 |
6 |
0 |
|
PD27 |
1.15 |
294.31 |
4 |
3 |
5 |
0 |
|
PD28 |
3.11 |
268.27 |
4 |
3 |
5 |
0 |
|
PD29 |
1.47 |
279.30 |
4 |
2 |
5 |
0 |
|
PD30 |
2.03 |
312.75 |
3 |
2 |
5 |
0 |
|
PD31 |
2.34 |
270.33 |
4 |
2 |
5 |
0 |
|
PD32 |
2.14 |
347.20 |
3 |
2 |
5 |
0 |
|
PD33 |
2.52 |
354.40 |
3 |
1 |
6 |
0 |
|
PD34 |
1.03 |
246.27 |
5 |
3 |
6 |
0 |
|
PD35 |
1.90 |
296.30 |
4 |
2 |
5 |
0 |
|
PD36 |
1.70 |
328.75 |
4 |
3 |
5 |
0 |
|
PD37 |
1.54 |
323.31 |
5 |
2 |
6 |
0 |
|
PD38 |
1.70 |
273.32 |
3 |
3 |
5 |
0 |
|
PD39 |
1.62 |
293.32 |
4 |
2 |
5 |
0 |
|
PD40 |
1.75 |
307.35 |
4 |
3 |
6 |
0 |
|
PD41 |
1.91 |
292.34 |
3 |
2 |
5 |
0 |
|
PD42 |
1.95 |
292.34 |
3 |
2 |
5 |
0 |
|
PD43 |
0.57 |
245.24 |
4 |
3 |
5 |
0 |
|
PD44 |
1.01 |
323.33 |
6 |
3 |
6 |
0 |
|
PD45 |
4.20 |
318.33 |
4 |
3 |
5 |
0 |
|
PD46 |
1.55 |
295.30 |
5 |
3 |
5 |
0 |
|
PD47 |
1.65 |
321.33 |
4 |
3 |
7 |
0 |
|
PD48 |
2.57 |
397.88 |
4 |
2 |
6 |
0 |
|
PD49 |
1.76 |
303.32 |
4 |
2 |
5 |
0 |
|
PD50 |
2.00 |
314.73 |
5 |
2 |
5 |
0 |
|
PD51 |
1.93 |
335.38 |
4 |
2 |
5 |
0 |
In-silico screening of toxicity prediction and molecular docking of the designed analogues:
Toxicity profile of novel proposed analogues was assessed using the Osiris Property Explorer and the ligands with novelty (0 identity), good drug-likeness properties and no toxicity were selected for molecular docking studies against JAK3 selected protein (PDB ID: 5toz) by Auto dock tool 2.0.
Table no 2: toxicity prediction and docking scores of the designed compounds
|
Compound Code |
Structure of the compounds |
Absence of toxicity |
Binding score |
|
PD1
|
|
yes |
-7.5 |
|
PD2 |
|
yes |
-7.77 |
|
PD3 |
|
yes |
-7.55 |
|
PD4 |
|
yes |
-6.61 |
|
PD5 |
|
yes |
-7.06 |
|
PD6 |
|
yes |
-6.48 |
|
PD7 |
|
yes |
-7.24 |
|
PD8 |
|
yes |
-7.32 |
|
PD9 |
|
yes |
-7.38 |
|
PD10 |
|
yes |
-8.35 |
|
PD11 |
|
yes |
-7.29 |
|
PD12 |
|
yes |
-7.43 |
|
PD13 |
|
yes |
-7.26 |
|
PD14 |
|
yes |
-8.52 |
|
PD15 |
|
yes |
-7.45 |
|
PD16 |
|
yes |
-7.49 |
|
PD17 |
|
yes |
-7.31 |
|
PD18 |
|
yes |
-7.33 |
|
PD19 |
|
yes |
-7.19 |
|
PD20 |
|
yes |
-7.93 |
|
PD21 |
|
yes |
-4.45 |
|
PD22 |
|
yes |
-7.6 |
|
PD23 |
|
yes |
-8.12 |
|
PD24 |
|
yes |
-7.82 |
|
PD25 |
|
yes |
-8.13 |
|
PD26 |
|
yes |
-8.22 |
|
PD27 |
|
yes |
-7.36 |
|
PD28 |
|
yes |
-7.72 |
|
PD29 |
|
yes |
-7.76 |
|
PD30 |
|
yes |
-7.61 |
|
PD31 |
|
yes |
-6.65 |
|
PD32 |
|
yes |
-7.9 |
|
PD33 |
|
yes |
-8.15 |
|
PD34 |
|
yes |
-7.94 |
|
PD35 |
|
yes |
-7.95 |
|
PD36 |
|
yes |
-7.94 |
|
PD37 |
|
yes |
-8.82 |
|
PD38 |
|
yes |
-6.97 |
|
PD39 |
|
yes |
-7.58 |
|
PD40 |
|
yes |
-7.89 |
|
PD41 |
|
yes |
-7.62 |
|
PD42 |
|
yes |
-6.78 |
|
PD43 |
|
yes |
-7.44 |
|
PD44 |
|
yes |
-7.66 |
|
PD45 |
|
yes |
-8.52 |
|
PD46 |
|
yes |
-7.69 |
|
PD47 |
|
yes |
-8.41 |
|
PD48 |
|
yes |
-5.9 |
|
PD49 |
|
yes |
-8.31 |
|
PD50 |
|
yes |
-7.74 |
|
PD51 |
|
yes |
-7.51 |
Table No.3: Structure / IUPAC name of top 5 ligands based on docking scores
|
Compound code |
Ligand structure |
IUPAC name |
|
PD23 |
|
2-anilino-N-(quinazolin-4-yl) acetamide
|
|
PD25 |
|
2-[(pyridin-4-yl) amino]-N-(quinazolin-4-yl) acetamide
|
|
PD26 |
|
2-(4-methoxyanilino)-N-(quinazolin-4-yl) acetamide |
|
PD37 |
|
2-(4-nitroanilino)-N-(quinazolin-4-yl) acetamide
|
|
PD45 |
|
2-[(1H-benzimidazol-2-yl) amino]-N-(quinazolin-4yl) acetamide
|
Post-Docking Analysis:
To visualize molecular structures and analyze interactions between protein and ligands, Molegro Molecular Viewer (MMV) software is used.
Table No.4: Docking interaction of top 5 ligands based on docking scores
|
Compound Code |
Interaction Of The Compounds |
|
PD23 |
|
|
PD25 |
|
|
PD26 |
|
|
PD37 |
|
|
PD45 |
|
CONCLUSION
This work illustrates the value of molecular docking and in silico drug design methods in locating novel quinozoline derivatives that may be used as JAK3 inhibitors to treat UC and CD. Strong binding affinities and advantageous pharmacokinetic profiles are suggested by the computational results, which makes these drugs attractive candidates for additional experimental verification. Improved outcomes for patients with UC and CD may result from the development of efficient and selective JAK3 targeted treatments made possible by the insights gathered from the docking analysis and SAR research
ACKNOWLEDGEMENTS
We express our sincere thanks to the Department of Pharmaceutical Chemistry, College of Pharmacy, Madras Medical College (MMC), Chennai for providing necessary facilities for the research work
Conflicts of Interest
The author declares there is no conflict of interest.
REFERENCES




Priyadharshini R*, Neelambari S., Jawaharsamuvel R., Mohammed Idrees H., Insilico Drug Design and Molecular Docking Studies of Some Novel Quinozoline Derivatives Targeting Janus Kinase 3 For Ulcerative Colitis and Crohn’s Diseases, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 01, 221-235. https://doi.org/10.5281/zenodo.14593394
 10.5281/zenodo.14593394
10.5281/zenodo.14593394




