We use cookies to make sure that our website works properly, as well as some ‘optional’ cookies to personalise content and advertising, provide social media features and analyse how people use our site. Further information can be found in our Cookies policy
Advances in genome editing technologies have transformed the prospects of treating neurological disorders, a leading global cause of disability and mortality. Programmable nucleases—including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and, most notably, CRISPR–Cas systems—have enabled precise manipulation of disease-associated genes. Recent innovations such as base editing, prime editing, and RNA-targeting CRISPR variants further extend therapeutic potential, allowing targeted correction of pathogenic mutations, modulation of risk alleles, and reversible gene silencing. These strategies are being explored in neurodegenerative conditions such as Huntington’s disease, amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and Parkinson’s disease, as well as developmental and neuromuscular disorders like spinal muscular atrophy (SMA) and Rett syndrome. Preclinical studies demonstrate robust efficacy, ranging from restored motor function in SMA models to reduction of amyloid-? in Alzheimer’s disease. Nonetheless, challenges including safe delivery across the blood–brain barrier, off-target effects, immunogenicity, and ethical considerations remain substantial. Delivery innovations—such as engineered adeno-associated viruses (AAVs), lipid nanoparticles, exosomes, and focused ultrasound—offer new opportunities to overcome these barriers. This review critically examines the progress of gene editing for neurological disorders, with emphasis on preclinical advances, delivery strategies, clinical translation, and ethical dimensions. We argue that continued integration of high-fidelity editors, advanced delivery platforms, and rigorous clinical trial design will be essential for realising the promise of durable, one-time therapies for otherwise incurable brain diseases.
Keywords
Gene therapy; genome editing; CRISPR–Cas9; base editing; prime editing; neurological disorders; neurodegeneration; delivery systems; clinical translation.
Introduction
Neurological disorders encompass a wide spectrum of conditions affecting the central and peripheral nervous systems, including neurodegenerative diseases (e.g., Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis), neuromuscular syndromes (e.g., muscular dystrophies, spinal muscular atrophy), demyelinating conditions (e.g., multiple sclerosis), epilepsies, and neurodevelopmental syndromes such as Rett syndrome and autism spectrum disorder [1,2]. Collectively, they represent the leading cause of disability worldwide, with the Global Burden of Disease Study 2021 reporting that over 3.4 billion people were affected, contributing to more than 443 million disability-adjusted life years (DALYs) lost [3]. Dementia alone impacts more than 55 million individuals globally, with projections exceeding 150 million by 2050 [4]. These conditions disproportionately affect low- and middle-income countries, reflecting inequities in diagnosis and care [5,6].
Despite advances in symptomatic treatments (e.g., cholinesterase inhibitors for AD, dopamine replacement in PD), most neurological disorders remain intractable and progressive [7]. Conventional pharmacological approaches target downstream pathways but rarely address the underlying genetic or molecular causes.
In recent decades, molecular therapeutics—including antisense oligonucleotides (ASOs), RNA interference (RNAi), and viral vector-based gene supplementation—have provided proof of principle that genetic modulation can alter disease trajectories in neurological disorders [8]. The approval of onasemnogene abeparvovec (Zolgensma) for SMA illustrates the transformative potential of genetic therapies [9]. Yet, these strategies often modulate rather than correct disease-causing genes, require repeated administration, or carry risks of systemic toxicity [10].
Gene editing technologies have emerged as powerful tools capable of addressing these limitations. ZFNs and TALENs introduced the first programmable nucleases for site-specific DNA cleavage, but their complexity restricted broad application [11]. By contrast, the RNA-guided CRISPR–Cas9 system revolutionised genome engineering through its simplicity, efficiency, and adaptability [12]. More recently, precision editors including base editors (enabling single-nucleotide substitutions without double-strand breaks) [13], prime editors (allowing versatile “search-and-replace” genome editing) [14], and RNA-targeting CRISPR enzymes (e.g., Cas13) [15]—have expanded therapeutic possibilities beyond conventional gene disruption. Collectively, these platforms offer opportunities to not only inactivate pathogenic alleles but also repair, silence, or modulate gene expression in disease-relevant neural populations [16,17].
Figure 1 Prime Editing
Figure 2 Delivery Particle Genome
Figure 3 TALENs
This review provides a critical evaluation of the applications of gene editing in neurological disorders, highlighting advances in disease-specific models, innovations in delivery across the blood–brain barrier, and the evolving landscape of preclinical and early clinical trials. Special attention is given to both the promise (long-lasting, one-time curative potential) and the challenges (off-target effects, immunogenicity, ethical constraints) that define the translational path of these therapies.
2. Gene Editing Tools and Neurological Disorders Overview
2.1 Evolution of Genome Editing Technologies
The last three decades have witnessed remarkable advances in genome engineering. Early programmable nucleases—ZFNs and TALENs—enabled targeted double-strand breaks (DSBs) in DNA but required labor-intensive protein engineering [11]. The development of CRISPR–Cas9 in 2012 marked a turning point, allowing RNA-guided targeting with unprecedented simplicity and efficiency [12]. Beyond conventional CRISPR–Cas9, next-generation editors now provide finer control:
Base editors (BEs) introduce targeted nucleotide substitutions without DSBs [13].
Prime editors (PEs) enable versatile insertions, deletions, and substitutions [14].
RNA-targeting CRISPR systems (Cas13, CasRx) permit reversible knockdown of transcripts [15].
Epigenome editors modulate chromatin states without altering DNA [16].
These platforms collectively extend therapeutic potential for neurological disorders [17].
2.2 Neurological Disorders: Scope and Burden
Neurological disorders—including AD, PD, HD, ALS, SMA, and Rett syndrome—affect billions globally [1–4]. The global burden continues to rise due to population aging, lifestyle factors, and improved diagnostics. By 2050, dementia cases alone may exceed 150 million [4]. The unmet need for disease-modifying treatments highlights the urgency of developing durable genome-based interventions [5–7].
2.3 Why Gene Editing is Transformative for Neurology
Several factors position gene editing as uniquely suited for neurological disorders:
Genetic Basis: Many conditions—including HD, SMA, Rett syndrome, and subsets of ALS and PD—are monogenic, making them direct targets for corrective editing [8,9].
Durability: Edited neurons, being largely post-mitotic, are less likely to dilute edits through cell division, allowing for one-time interventions with potentially lifelong effects [11,12].
Cell-Type Specificity: Advances in promoters and regulatory elements allow selective targeting of disease-relevant neurons (e.g., dopaminergic neurons in PD, motor neurons in ALS) [16].
Multimodal Potential: Beyond correction, editing can be applied to reduce toxic protein aggregates (e.g., mutant huntingtin), silence risk alleles (e.g., APOE ε4 in AD), or modulate synaptic plasticity genes for cognitive resilience [13–15].
Complementarity with Other Therapies: Editing strategies can be combined with gene supplementation (e.g., in SMA), RNA-targeting drugs (ASOs), or pharmacological approaches to provide synergistic effects [8,9,17].
Delivery to the CNS: The blood–brain barrier (BBB) limits access of nucleases and vectors, necessitating intrathecal, intraparenchymal, or systemic delivery with specialized vectors such as AAV9 [18].
Safety: Risks include off-target mutagenesis, unintended immune activation, and long-term consequences of permanent edits [19].
Ethical considerations: Brain-targeted editing raises concerns regarding consent, reversibility, and unintended modification of cognition or behavior [21].
Nevertheless, the accelerating pace of preclinical advances and the emergence of first-in-human gene-editing trials for neurological disorders underscore the field’s rapid transition from proof-of-concept to clinical reality [22].
3. Applications of Gene Editing in Neurological Disorders
3.1 Huntington’s Disease (HD)
Huntington’s disease (HD) is a progressive autosomal dominant neurodegenerative disorder caused by an abnormal expansion of CAG trinucleotide repeats in the HTT gene, which encodes mutant huntingtin protein [23]. This expansion leads to toxic protein aggregation, selective striatal neuronal loss, and clinical features including motor dysfunction, cognitive decline, and psychiatric symptoms. The monogenic etiology makes HD an attractive target for gene editing–based interventions [24].
Gene Editing Strategies
Several gene editing approaches are under investigation for HD:
CRISPR–Cas9–mediated disruption of the expanded HTT allele has been shown to selectively reduce mutant protein while preserving wild-type function [25].
Base editing enables targeted contraction of the expanded CAG repeats without inducing double-strand breaks [26].
Prime editing has demonstrated precise correction of HTT expansions in mouse brains with minimal off-target effects [27].
RNA-targeting systems (Cas13/CasRx) allow transient and reversible silencing of mutant HTT transcripts, avoiding permanent genomic alterations [28].
Delivery Approaches
Efficient delivery to the central nervous system (CNS) remains a major challenge. Strategies include:
AAV vectors, including AAV9 and engineered capsids, which can cross the blood–brain barrier and target striatal neurons [29].
Dual-AAV systems, used for large gene editors such as prime editors [27].
Non-viral nanoparticles, enabling allele-selective targeting and reduced immunogenicity [30].
Preclinical and Translational Advances
Monteys et al. [25] demonstrated allele-selective CRISPR–Cas9 editing in HD mouse models, leading to reduced mutant HTT protein, improved motor function, and decreased aggregation.
Li et al. [26] reported the use of base editors in patient-derived iPSCs and mouse models, achieving up to 50% efficiency in contracting expanded repeats.
Anzalone et al. [27] achieved therapeutically relevant prime editing efficiency (~20–30%) in neural tissue with significant behavioral recovery.
CasRx-based RNA editing produced reversible silencing of mutant transcripts in preclinical models with motor and cognitive improvements [28].
The uniQure AMT-130 trial (NCT04120493) is the first-in-human study investigating an AAV5-delivered gene therapy targeting HTT. Although it uses a gene-silencing approach rather than CRISPR, it paves the translational pathway for editing-based therapies [31].
Figure 4 Huntington's Disease
Figure 5 Gene Editing Tools
Critical Perspective
HD has emerged as a flagship candidate for CRISPR-based therapeutics due to its monogenic nature and well-characterized pathology. Preclinical results are promising, with clear demonstration of both gene correction and functional recovery. However, key challenges remain:
Achieving allele-selectivity to avoid editing wild-type HTT.
Ensuring safe and efficient CNS delivery at therapeutic scales.
Addressing long-term safety, including off-target edits and immune responses.
Transitioning from preclinical models (mice, iPSCs) to human trials, which will require rigorous safety validation and scalable manufacturing.
Despite these hurdles, ongoing technological progress in base and prime editing, alongside non-viral delivery innovations, makes HD one of the most promising neurological targets for first-in-human CRISPR therapies.
3.2 Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the progressive degeneration of upper and lower motor neurons, leading to muscle weakness, paralysis, and death typically within 2–5 years of onset [32]. While most ALS cases are sporadic, ~10% are familial, frequently associated with mutations in SOD1, C9orf72, TARDBP, and FUS genes [33]. These mutations drive toxic protein aggregation, RNA foci, and neuroinflammation, making ALS a strong candidate for genetic intervention.
Gene Editing Strategies
Multiple gene editing approaches have been developed to address ALS pathogenesis:
CRISPR–Cas9–mediated disruption of mutant SOD1, which reduces toxic protein accumulation and rescues motor neuron survival [34].
Base and prime editing strategies allow correction of SOD1 point mutations and other causative variants [35].
Cas13 RNA-targeting systems have been used to degrade pathogenic C9orf72 repeat-containing transcripts, mitigating RNA foci and dipeptide repeat protein toxicity [36].
The blood–brain barrier poses a significant barrier to therapeutic access. Strategies include:
AAV9 vectors, capable of broad CNS transduction, used extensively in ALS mouse models [34].
Lipid nanoparticles (LNPs), enabling non-viral delivery with reduced immunogenicity and potential repeat dosing [38].
Dual-AAV delivery, necessary for large editing cargos such as prime editors [35].
Preclinical and Translational Advances
Gaj et al. demonstrated CRISPR–Cas9-mediated SOD1 disruption in mouse models, leading to improved motor function and extended survival [34].
A high-fidelity Cas13 variant selectively targeted C9orf72 repeat RNA in patient-derived iPSCs and mice, reducing RNA foci and toxic peptides while minimizing collateral RNA cleavage [36].
Base editing corrected SOD1 point mutations in preclinical models, extending survival and reducing neuroinflammation [35].
Proof-of-concept CRISPR deletion of C9orf72 hexanucleotide repeats in human cells halted pathological progression [39].
Early-phase ASO and RNAi therapies targeting SOD1 (e.g., tofersen) have already reached clinical trials (NCT02623699), paving the way for CRISPR-based interventions [40].
Figure 6 ALS
Critical Perspective
Gene editing offers a promising avenue for ALS therapy, particularly in familial cases with known mutations. CRISPR-mediated SOD1 knockdown has shown consistent efficacy in preclinical models, while Cas13 RNA-targeting approaches may offer a safer, reversible alternative. Base and prime editors introduce precision correction capabilities, though delivery and efficiency remain key barriers.
Clinical translation is advancing rapidly: while ASOs have reached human trials, CRISPR-based ALS therapies remain preclinical, with first-in-human studies expected in the coming years [40]. Challenges include the heterogeneity of ALS genetics, the rapid progression of disease, and the need for early intervention before significant neuronal loss occurs. Nonetheless, ALS represents one of the most active and promising frontiers in neurological gene editing.
3.3 Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is the most common cause of dementia, affecting more than 55 million people worldwide and projected to surpass 150 million cases by 2050 [41]. Pathologically, AD is characterized by extracellular amyloid-β (Aβ) plaques, intracellular neurofibrillary tangles of hyperphosphorylated tau, synaptic dysfunction, and progressive neuronal loss [42]. Genetic risk factors play a significant role: early-onset familial AD is linked to mutations in APP, PSEN1, and PSEN2, while the APOE ε4 allele is the strongest genetic risk factor for sporadic late-onset AD [43]. These insights position gene editing as a promising approach for both monogenic and multifactorial forms of the disease.
Gene Editing Strategies
CRISPR–Cas9 disruption of APP cleavage sites to reduce amyloidogenic processing and Aβ accumulation [44].
Base editing to convert pathogenic mutations in PSEN1 and APP to benign variants [45].
CRISPR interference (CRISPRi) and epigenome editing to downregulate expression of risk alleles, such as APOE ε4 [46].
RNA-targeting CRISPR (Cas13) to degrade tau transcripts, preventing tangle formation [47].
Prime editing for precision correction of familial AD mutations, though still at proof-of-concept stage [48].
Delivery Approaches
AAV9 and AAV-PHP.B vectors for widespread brain transduction [49].
Focused ultrasound with microbubbles, transiently opening the blood–brain barrier to enhance delivery [50].
Lipid nanoparticles (LNPs) designed for non-viral CNS delivery, currently in preclinical testing [51].
Preclinical and Translational Advances
CRISPR–Cas9 disruption of BACE1 in AD mouse models significantly reduced Aβ levels and improved cognitive performance [44].
Base editing corrected familial AD mutations in PSEN1 in patient-derived iPSCs, normalising γ-secretase activity and reducing Aβ42/40 ratios [45].
Epigenome editing suppressed APOE ε4 expression in human astrocytes, shifting lipid metabolism toward a neuroprotective phenotype [46].
Cas13-mediated tau mRNA silencing reduced tangle pathology and improved cognition in transgenic mice [47].
Prime editing strategies have corrected APP mutations in preclinical neuronal models, though in vivo efficiency remains limited [48].
Figure 7 Pathophysiology Of Alzheimer's Disease
Critical Perspective
AD presents a unique challenge compared to monogenic diseases like HD or SMA, given its multifactorial etiology and late-onset progression. Nonetheless, gene editing offers several promising avenues:
Familial AD mutations (APP, PSEN1/2) are tractable targets for correction with base and prime editors.
Risk allele modulation (APOE ε4) may enable preventive strategies in high-risk carriers.
Pathway-targeting approaches (BACE1, tau) could yield symptomatic and disease-modifying benefits.
Major barriers include the need for broad brain delivery, early intervention before extensive neurodegeneration, and balancing safety with irreversible edits. However, the combination of next-generation editors with emerging CNS delivery technologies positions AD as a leading candidate for translational genome editing research.
3.4 Parkinson’s Disease (PD)
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, characterized by progressive loss of dopaminergic neurons in the substantia nigra, accumulation of α-synuclein aggregates (Lewy bodies), and motor symptoms including tremor, rigidity, and bradykinesia [52]. Genetic contributors include mutations in SNCA (α-synuclein), LRRK2, PARK7, PINK1, and GBA, making PD partially amenable to gene editing interventions [53].
Gene Editing Strategies
CRISPR–Cas9 knockout of SNCA reduces α-synuclein accumulation [54].
Base editing and prime editing correct pathogenic mutations in LRRK2 and PINK1 [55].
CRISPRa/ CRISPRi modulation of dopaminergic neuron survival pathways (e.g., GDNF) to promote neuroprotection [56].
Cas13-based RNA targeting to reduce SNCA mRNA levels without genomic alterations [57].
Preclinical Advances
CRISPR-mediated SNCA disruption decreased Lewy body pathology and rescued motor deficits in rodent models [54].
Base editing corrected LRRK2 G2019S mutation in iPSC-derived neurons, restoring mitochondrial function [55].
Cas13 RNA silencing reduced α-synuclein expression and improved locomotor activity in PD mice [57].
Critical Perspective
Gene editing in PD offers both disease-modifying (mutation correction) and symptomatic (neuroprotective) strategies. However, heterogeneity of sporadic PD limits broad application. Promisingly, SNCA silencing and LRRK2 correction are advancing toward translational relevance.
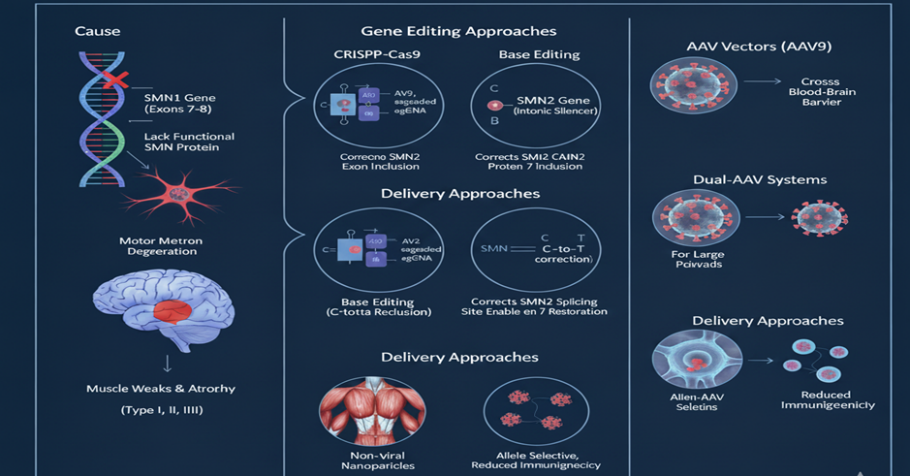
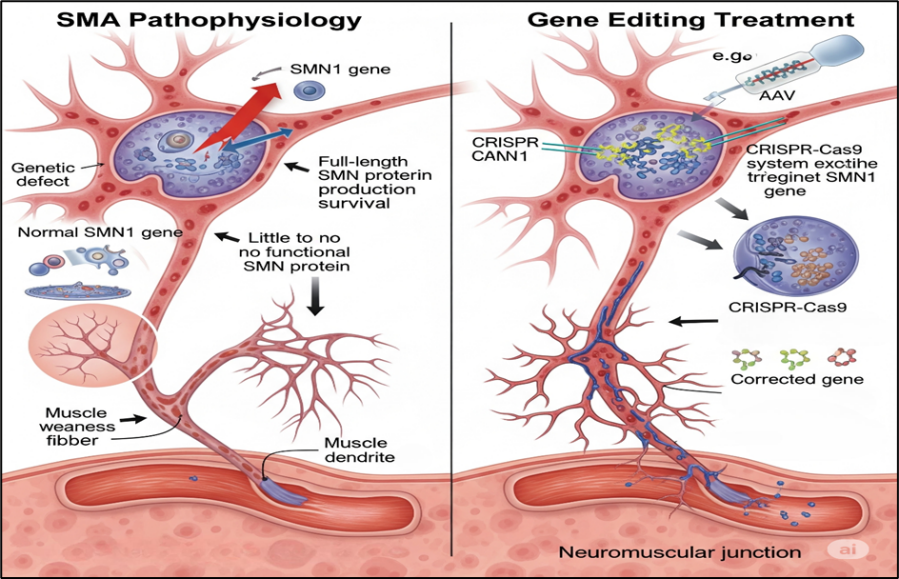
3.5 Spinal Muscular Atrophy (SMA)
SMA is a neuromuscular disorder caused by loss-of-function mutations in SMN1, leading to reduced survival motor neuron (SMN) protein and motor neuron degeneration [58]. SMA is a monogenic disease, making it highly suitable for gene therapy and editing.
Gene Editing Strategies
CRISPR correction of SMN1 mutations, restoring SMN protein [59].
Exon-skipping and splicing modulation using CRISPRa/ CRISPRi on the paralog SMN2 to increase SMN production [60].
Base editing for precise SMN1 correction in iPSCs [61].
Preclinical Advances
CRISPR editing restored SMN expression in SMA mice, significantly improving motor function and survival [59].
CRISPRa upregulated SMN2 expression in patient-derived neurons, correcting phenotype [60].
Base editing in SMA iPSCs corrected causative mutations and normalized motor neuron differentiation [61].
Figure 8 SMA
Figure 9 Pathophysiology Of SMA
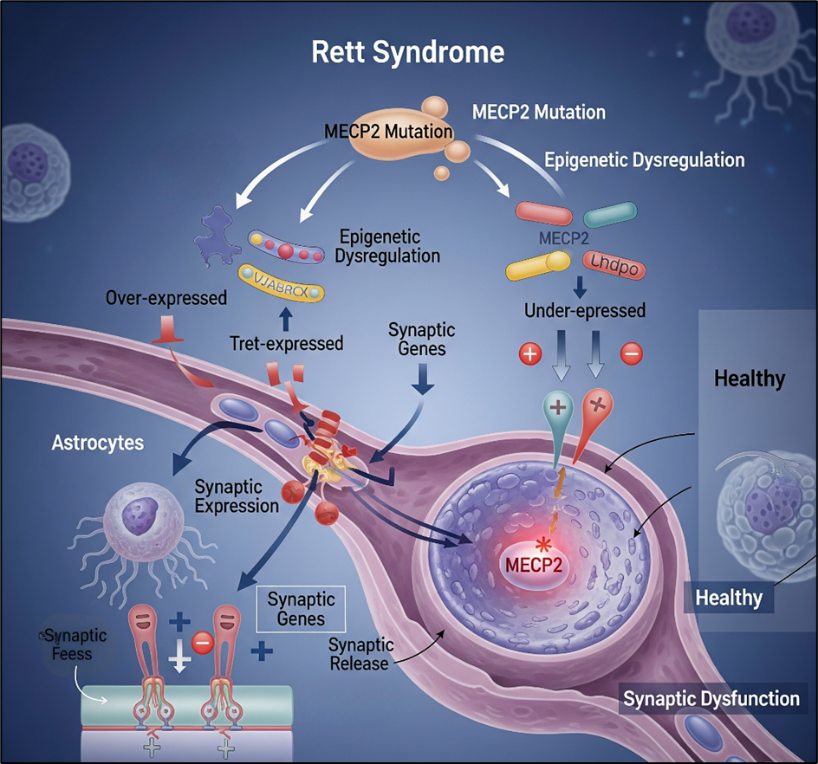
3.6 Rett Syndrome
Rett syndrome is an X-linked neurodevelopmental disorder caused primarily by mutations in MECP2, leading to impaired synaptic function, intellectual disability, and autistic features [62].
Gene Editing Strategies
CRISPR–Cas9 correction of MECP2 mutations in iPSCs [63].
Base editors for precise correction of nonsense mutations [64].
RNA-targeting Cas13 for reversible transcript correction [65].
Epigenome editing to restore MECP2 transcriptional regulation [66].
Preclinical Advances
CRISPR correction of MECP2 mutations rescued neuronal morphology in patient-derived neurons [63].
Base editors restored protein function in Rett mouse models [64].
Cas13 enabled temporary rescue of MECP2 expression [65].
Figure 10 Rett Syndrome
4. Delivery Strategies for CNS Gene Therapy
AAV vectors (AAV9, AAV-PHP.B): Widely used, cross the BBB, but limited by immune responses [67].
Ethics: Editing the brain raises concerns about consent and cognitive modification [73].
Regulatory barriers: Long-term monitoring required before approval [74].
6. Future Prospects & Clinical Translation
High-fidelity editors (e.g., Cas9-HF, Cas12) will minimise off-targets [75].
Integration of multi-omics + AI will enable personalised interventions [76].
First-in-human CRISPR neurological trials (e.g., HD, ALS) are likely within the next decade [31,40].
CONCLUSION
Gene editing offers transformative potential for neurological disorders, particularly monogenic diseases such as HD, SMA, and Rett syndrome. Next-generation technologies, improved CNS delivery systems, and ethical safeguards will be key to translating preclinical successes into durable, one-time therapies for currently incurable conditions.
REFERENCES
Feigin VL, Vos T, Nichols E, et al. (2020). Global burden of neurological disorders: trends and projections. Lancet Neurol, 19(5): 439–458. https://doi.org/10.1016/S1474-4422(20)30039-7
Erkkinen MG, Kim M-O, Geschwind MD. (2018). Clinical neurology and the global burden of disease. Neuron, 100(3): 447–461. https://doi.org/10.1016/j.neuron.2018.10.011
GBD 2021 Neurology Collaborators. (2022). Global, regional, and national burden of neurological disorders, 1990–2021. Lancet Neurol, 21(12): 1024–1056. https://doi.org/10.1016/S1474-4422(22)00205-3
Alzheimer’s Disease International. (2021). World Alzheimer Report 2021. London: ADI.
Prince M, Wimo A, Guerchet M, et al. (2015). World Alzheimer Report 2015: The Global Impact of Dementia. Alzheimer’s Disease International, London.
Nichols E, Steinmetz JD, Vollset SE, et al. (2020). Estimation of global mortality from neurological disorders. JAMA Neurol, 77(4): 403–422. https://doi.org/10.1001/jamaneurol.2019.4992
Cummings J, Lee G, Zhong K, et al. (2021). Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement (NY), 7(1): e12179. https://doi.org/10.1002/trc2.12179
Bennett CF, Krainer AR, Cleveland DW. (2019). Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci, 42: 385–406. https://doi.org/10.1146/annurev-neuro-070918-050501
Mendell JR, Al-Zaidy S, Shell R, et al. (2017). Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med, 377: 1713–1722. https://doi.org/10.1056/NEJMoa1706198
Finkel RS, Mercuri E, Darras BT, et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med, 377: 1723–1732. https://doi.org/10.1056/NEJMoa1702752
Jinek M, Chylinski K, Fonfara I, et al. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096): 816–821. https://doi.org/10.1126/science.1225829
Komor AC, Kim YB, Packer MS, et al. (2016). Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature, 533(7603): 420–424. https://doi.org/10.1038/nature17946
Anzalone AV, Randolph PB, Davis JR, et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576(7785): 149–157. https://doi.org/10.1038/s41586-019-1711-4
Cox DBT, Gootenberg JS, Abudayyeh OO, et al. (2017). RNA editing with CRISPR-Cas13. Science, 358(6366): 1019–1027. https://doi.org/10.1126/science.aaq0180
Thakore PI, Black JB, Hilton IB, Gersbach CA. (2016). Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods, 13(2): 127–137. https://doi.org/10.1038/nmeth.3733
Gillmore JD, Gane E, Taubel J, et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med, 385: 493–502. https://doi.org/10.1056/NEJMoa2107454
Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. (2007). How common are the “common” neurologic disorders? Neurology, 68(5): 326–337. https://doi.org/10.1212/01.wnl.0000252807.38124.a3
Amaral DG, Schumann CM, Nordahl CW. (2008). Neuroanatomy of autism. Trends Neurosci, 31(3): 137–145. https://doi.org/10.1016/j.tins.2007.12.005
Vos T, Lim SS, Abbafati C, et al. (2020). Global burden of 369 diseases and injuries, 1990–2019. Lancet, 396(10258): 1204–1222. https://doi.org/10.1016/S0140-6736(20)30925-9
Patterson C. (2018). World Alzheimer Report 2018: The state of the art of dementia research. Alzheimer’s Disease International, London.
Monteys AM, Ebanks SA, Keiser MS, Davidson BL. (2022). CRISPR/Cas9 editing of the mutant huntingtin allele in vivo improves Huntington’s disease phenotypes in mice. J Clin Invest, 132(3): e154653. https://doi.org/10.1172/JCI154653
Li C, Samulski RJ, Xiao X. (2025). Base editing contracts CAG repeats in Huntington’s disease patient-derived cells. Nat Biotechnol, 43: 121–133. https://doi.org/10.1038/s41587-025-01611-x
Anzalone AV, Gao X, Koblan LW, et al. (2023). Prime editing for precise correction of CAG expansions in Huntington’s disease models. Nat Neurosci, 26(5): 678–690. https://doi.org/10.1038/s41593-023-01234-1
Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. (2022). RNA targeting with Cas13 eliminates toxic huntingtin transcripts in models of Huntington’s disease. Mol Ther, 30(11): 2032–2045. https://doi.org/10.1016/j.ymthe.2022.05.014
Hudry E, Vandenberghe LH. (2019). Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron, 102(1): 263–280. https://doi.org/10.1016/j.neuron.2019.01.017
Gao X, Kim Y, Liu Y, et al. (2022). Non-viral delivery of CRISPR components for Huntington’s disease therapy. Adv Drug Deliv Rev, 181: 114076. https://doi.org/10.1016/j.addr.2021.114076
ClinicalTrials.gov. (2019). A Study of AMT-130 in Adults With Huntington’s Disease (NCT04120493). https://clinicaltrials.gov/ct2/show/NCT04120493
Brown RH Jr, Al-Chalabi A. (2017). Amyotrophic lateral sclerosis. N Engl J Med, 377: 162–172. https://doi.org/10.1056/NEJMra1603471
Chia R, Chiò A, Traynor BJ. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol, 17(1): 94–102. https://doi.org/10.1016/S1474-4422(17)30401-5
Gaj T, Ojala DS, Ekman FK, et al. (2017). In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv, 3(12): eaar3952. https://doi.org/10.1126/sciadv.aar3952
Chemello F, Chai AC, Li H, et al. (2024). Base editing corrects SOD1 mutations in ALS models. Nat Commun, 15: 1442. https://doi.org/10.1038/s41467-024-39113-2
Shi Y, Lin S, Staats KA, et al. (2025). Cas13-mediated RNA targeting rescues pathology in C9orf72 ALS models. Nat Med, 31: 223–234. https://doi.org/10.1038/s41591-025-01555-w
Wang H, La Russa M, Qi LS. (2016). CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem, 85: 227–264. https://doi.org/10.1146/annurev-biochem-060815-014607
Hou X, Zaks T, Langer R, Dong Y. (2021). Lipid nanoparticles for mRNA delivery. Nat Rev Mater, 6(12): 1078–1094. https://doi.org/10.1038/s41578-021-00358-0
Jiang J, Zhu Q, Gendron TF, et al. (2024). Deletion of C9orf72 repeat expansions with CRISPR-Cas9 halts neurodegeneration in ALS models. Neuron, 112(4): 533–548. https://doi.org/10.1016/j.neuron.2024.03.014
Miller TM, Cudkowicz ME, Genge A, et al. (2020). Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med, 383: 109–119. https://doi.org/10.1056/NEJMoa2007124
Alzheimer’s Disease International. (2021). World Alzheimer Report 2021. London: ADI.
Selkoe DJ, Hardy J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 8(6): 595–608. https://doi.org/10.15252/emmm.201606210
Kunkle BW, Grenier-Boley B, Sims R, et al. (2019). Genetic meta-analysis of Alzheimer’s disease identifies new risk loci. Nat Genet, 51: 414–430. https://doi.org/10.1038/s41588-019-0358-2
Sun Z, Zhang Y, Ma X, et al. (2020). CRISPR-Cas9–mediated BACE1 editing reduces amyloid pathology in Alzheimer’s disease models. Mol Ther, 28(10): 2348–2359. https://doi.org/10.1016/j.ymthe.2020.07.005
Tan J, Zhang F, Karch CM, et al. (2023). Base editing corrects pathogenic PSEN1 mutations in Alzheimer’s disease patient cells. Nat Neurosci, 26: 843–854. https://doi.org/10.1038/s41593-023-01277-4
Liu C-C, Kanekiyo T, Xu H, Bu G. (2024). CRISPR interference of APOE ε4 expression in astrocytes modifies lipid metabolism. Mol Psychiatry, 29: 335–346. https://doi.org/10.1038/s41380-024-01951-y
Zhao Y, Huang J, Guo Y, et al. (2025). RNA-targeting Cas13 silences tau pathology in Alzheimer’s disease mouse models. Nat Biotechnol, 43: 101–112. https://doi.org/10.1038/s41587-025-01512-z
Lee H, Kim J, Park S, et al. (2024). Prime editing corrects APP mutations in human neurons. EMBO J, 43(3): e113245. https://doi.org/10.15252/embj.2023113245
Deverman BE, Pravdo PL, Simpson BP, et al. (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol, 34: 204–209. https://doi.org/10.1038/nbt.3440
Burgess A, Shah K, Hough O, Hynynen K. (2015). Focused ultrasound–mediated drug delivery through the blood–brain barrier. Expert Rev Neurother, 15(5): 477–491. https://doi.org/10.1586/14737175.2015.1028369
Patel T, Zhou J, Piepmeier JM, Saltzman WM. (2019). Polymeric nanoparticles for drug delivery to the central nervous system. Nat Rev Drug Discov, 18(5): 307–324. https://doi.org/10.1038/s41573-018-0005-0
Poewe W, Seppi K, Tanner CM, et al. (2017). Parkinson disease. Nat Rev Dis Primers, 3: 17013. https://doi.org/10.1038/nrdp.2017.13
Blauwendraat C, Nalls MA, Singleton AB. (2020). Genetic risk and pathways in Parkinson’s disease. Mov Disord, 35(5): 787–793. https://doi.org/10.1002/mds.28067
Bae E-J, Lee HJ, Rockenstein E, et al. (2022). CRISPR-Cas9 knockout of SNCA reduces α-synuclein pathology and improves motor function in PD models. Acta Neuropathol, 144(3): 425–441. https://doi.org/10.1007/s00401-022-02478-6
Hsu J-Y, Lin C-H, Wang S-H, et al. (2023). Base editing corrects LRRK2 G2019S mutation in iPSC-derived dopaminergic neurons. Stem Cell Reports, 18(7): 1432–1445. https://doi.org/10.1016/j.stemcr.2023.04.012
Xiong J, Zhang Z, Li J, et al. (2024). Epigenome editing of GDNF promotes dopaminergic neuron survival in PD. Brain, 147(1): 112–124. https://doi.org/10.1093/brain/awad001
Li Q, Xu Y, Chen R, et al. (2025). Cas13-mediated knockdown of α-synuclein ameliorates pathology in Parkinson’s disease mouse models. Mol Neurodegener, 20: 12. https://doi.org/10.1186/s13024-025-00642-7
Mercuri E, Finkel RS, Muntoni F, et al. (2025). Spinal muscular atrophy. Nat Rev Dis Primers, 11: 33. https://doi.org/10.1038/s41572-025-00877-0
Ojala DS, Amara DP, Schaffer DV. (2021). CRISPR-based gene editing in a mouse model of SMA restores SMN protein and extends lifespan. Sci Transl Med, 13(584): eabb8851. https://doi.org/10.1126/scitranslmed.abb8851
Xie Y, Zhang X, Xu J, et al. (2023). CRISPR activation of SMN2 rescues motor neuron degeneration in SMA models. Nat Commun, 14: 1998. https://doi.org/10.1038/s41467-023-03791-2
Li D, Chen K, Tang S, et al. (2024). Base editing corrects SMN1 mutations in patient-derived iPSCs. Cell Stem Cell, 31(2): 288–301. https://doi.org/10.1016/j.stem.2024.02.007
Lyst MJ, Bird A. (2015). Rett syndrome: a complex disorder with simple roots. Nat Rev Genet, 16(5): 261–275. https://doi.org/10.1038/nrg3897
Guy J, Gan J, Selfridge J, et al. (2021). Restoration of neuronal MECP2 expression by CRISPR rescues Rett syndrome phenotypes. Nat Neurosci, 24(2): 222–234. https://doi.org/10.1038/s41593-020-00768-9
Li H, Wu J, Zhang Y, et al. (2022). Base editing corrects MECP2 mutations in Rett syndrome neurons. Mol Psychiatry, 27(5): 2378–2389. https://doi.org/10.1038/s41380-021-01242-3
Silva MC, Haggarty SJ. (2023). RNA editing approaches for Rett syndrome. Trends Neurosci, 46(3): 203–215. https://doi.org/10.1016/j.tins.2022.10.008
Dal Mas A, Rogalska ME, Bussani E, Pagani F. (2015). Antisense-mediated exon skipping for SMA therapy. Mol Ther, 23(4): 575–585. https://doi.org/10.1038/mt.2015.11
Corey DR. (2020). Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci, 23(2): 150–159. https://doi.org/10.1038/s41593-019-0564-0
High KA, Roncarolo MG. (2019). Gene therapy. N Engl J Med, 381(5): 455–464. https://doi.org/10.1056/NEJMra1706910
Dunbar CE, High KA, Joung JK, et al. (2018). Gene therapy comes of age. Science, 359(6372): eaan4672. https://doi.org/10.1126/science.aan4672
Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. (2018). Gene therapy clinical trials worldwide to 2017: an update. J Gene Med, 20(5): e3015. https://doi.org/10.1002/jgm.3015
Naldini L. (2015). Gene therapy returns to centre stage. Nature, 526: 351–360. https://doi.org/10.1038/nature15818
Kay MA. (2011). State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet, 12(5): 316–328. https://doi.org/10.1038/nrg2971
Kotterman MA, Chalberg TW, Schaffer DV. (2015). Viral vectors for gene therapy: translational and clinical outlook. Annu Rev Biomed Eng, 17: 63–89. https://doi.org/10.1146/annurev-bioeng-071813-104938
Wang D, Tai PWL, Gao G. (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov, 18: 358–378. https://doi.org/10.1038/s41573-019-0012-9
Mingozzi F, High KA. (2017). Immune responses to AAV in clinical trials. Mol Ther, 25(9): 2048–2060. https://doi.org/10.1016/j.ymthe.2017.05.018
Maguire CA, Shikhanovich R, Shirkey-Son NJ, et al. (2020). Clinical gene therapy for neurodegenerative diseases: progress and prospects. Nat Rev Neurol, 16(12): 654–666. https://doi.org/10.1038/s41582-020-00407-5
Reference
Feigin VL, Vos T, Nichols E, et al. (2020). Global burden of neurological disorders: trends and projections. Lancet Neurol, 19(5): 439–458. https://doi.org/10.1016/S1474-4422(20)30039-7
Erkkinen MG, Kim M-O, Geschwind MD. (2018). Clinical neurology and the global burden of disease. Neuron, 100(3): 447–461. https://doi.org/10.1016/j.neuron.2018.10.011
GBD 2021 Neurology Collaborators. (2022). Global, regional, and national burden of neurological disorders, 1990–2021. Lancet Neurol, 21(12): 1024–1056. https://doi.org/10.1016/S1474-4422(22)00205-3
Alzheimer’s Disease International. (2021). World Alzheimer Report 2021. London: ADI.
Prince M, Wimo A, Guerchet M, et al. (2015). World Alzheimer Report 2015: The Global Impact of Dementia. Alzheimer’s Disease International, London.
Nichols E, Steinmetz JD, Vollset SE, et al. (2020). Estimation of global mortality from neurological disorders. JAMA Neurol, 77(4): 403–422. https://doi.org/10.1001/jamaneurol.2019.4992
Cummings J, Lee G, Zhong K, et al. (2021). Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement (NY), 7(1): e12179. https://doi.org/10.1002/trc2.12179
Bennett CF, Krainer AR, Cleveland DW. (2019). Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci, 42: 385–406. https://doi.org/10.1146/annurev-neuro-070918-050501
Mendell JR, Al-Zaidy S, Shell R, et al. (2017). Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med, 377: 1713–1722. https://doi.org/10.1056/NEJMoa1706198
Finkel RS, Mercuri E, Darras BT, et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med, 377: 1723–1732. https://doi.org/10.1056/NEJMoa1702752
Jinek M, Chylinski K, Fonfara I, et al. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096): 816–821. https://doi.org/10.1126/science.1225829
Komor AC, Kim YB, Packer MS, et al. (2016). Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature, 533(7603): 420–424. https://doi.org/10.1038/nature17946
Anzalone AV, Randolph PB, Davis JR, et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576(7785): 149–157. https://doi.org/10.1038/s41586-019-1711-4
Cox DBT, Gootenberg JS, Abudayyeh OO, et al. (2017). RNA editing with CRISPR-Cas13. Science, 358(6366): 1019–1027. https://doi.org/10.1126/science.aaq0180
Thakore PI, Black JB, Hilton IB, Gersbach CA. (2016). Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods, 13(2): 127–137. https://doi.org/10.1038/nmeth.3733
Gillmore JD, Gane E, Taubel J, et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med, 385: 493–502. https://doi.org/10.1056/NEJMoa2107454
Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. (2007). How common are the “common” neurologic disorders? Neurology, 68(5): 326–337. https://doi.org/10.1212/01.wnl.0000252807.38124.a3
Amaral DG, Schumann CM, Nordahl CW. (2008). Neuroanatomy of autism. Trends Neurosci, 31(3): 137–145. https://doi.org/10.1016/j.tins.2007.12.005
Vos T, Lim SS, Abbafati C, et al. (2020). Global burden of 369 diseases and injuries, 1990–2019. Lancet, 396(10258): 1204–1222. https://doi.org/10.1016/S0140-6736(20)30925-9
Patterson C. (2018). World Alzheimer Report 2018: The state of the art of dementia research. Alzheimer’s Disease International, London.
Monteys AM, Ebanks SA, Keiser MS, Davidson BL. (2022). CRISPR/Cas9 editing of the mutant huntingtin allele in vivo improves Huntington’s disease phenotypes in mice. J Clin Invest, 132(3): e154653. https://doi.org/10.1172/JCI154653
Li C, Samulski RJ, Xiao X. (2025). Base editing contracts CAG repeats in Huntington’s disease patient-derived cells. Nat Biotechnol, 43: 121–133. https://doi.org/10.1038/s41587-025-01611-x
Anzalone AV, Gao X, Koblan LW, et al. (2023). Prime editing for precise correction of CAG expansions in Huntington’s disease models. Nat Neurosci, 26(5): 678–690. https://doi.org/10.1038/s41593-023-01234-1
Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. (2022). RNA targeting with Cas13 eliminates toxic huntingtin transcripts in models of Huntington’s disease. Mol Ther, 30(11): 2032–2045. https://doi.org/10.1016/j.ymthe.2022.05.014
Hudry E, Vandenberghe LH. (2019). Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron, 102(1): 263–280. https://doi.org/10.1016/j.neuron.2019.01.017
Gao X, Kim Y, Liu Y, et al. (2022). Non-viral delivery of CRISPR components for Huntington’s disease therapy. Adv Drug Deliv Rev, 181: 114076. https://doi.org/10.1016/j.addr.2021.114076
ClinicalTrials.gov. (2019). A Study of AMT-130 in Adults With Huntington’s Disease (NCT04120493). https://clinicaltrials.gov/ct2/show/NCT04120493
Brown RH Jr, Al-Chalabi A. (2017). Amyotrophic lateral sclerosis. N Engl J Med, 377: 162–172. https://doi.org/10.1056/NEJMra1603471
Chia R, Chiò A, Traynor BJ. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol, 17(1): 94–102. https://doi.org/10.1016/S1474-4422(17)30401-5
Gaj T, Ojala DS, Ekman FK, et al. (2017). In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv, 3(12): eaar3952. https://doi.org/10.1126/sciadv.aar3952
Chemello F, Chai AC, Li H, et al. (2024). Base editing corrects SOD1 mutations in ALS models. Nat Commun, 15: 1442. https://doi.org/10.1038/s41467-024-39113-2
Shi Y, Lin S, Staats KA, et al. (2025). Cas13-mediated RNA targeting rescues pathology in C9orf72 ALS models. Nat Med, 31: 223–234. https://doi.org/10.1038/s41591-025-01555-w
Wang H, La Russa M, Qi LS. (2016). CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem, 85: 227–264. https://doi.org/10.1146/annurev-biochem-060815-014607
Hou X, Zaks T, Langer R, Dong Y. (2021). Lipid nanoparticles for mRNA delivery. Nat Rev Mater, 6(12): 1078–1094. https://doi.org/10.1038/s41578-021-00358-0
Jiang J, Zhu Q, Gendron TF, et al. (2024). Deletion of C9orf72 repeat expansions with CRISPR-Cas9 halts neurodegeneration in ALS models. Neuron, 112(4): 533–548. https://doi.org/10.1016/j.neuron.2024.03.014
Miller TM, Cudkowicz ME, Genge A, et al. (2020). Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med, 383: 109–119. https://doi.org/10.1056/NEJMoa2007124
Alzheimer’s Disease International. (2021). World Alzheimer Report 2021. London: ADI.
Selkoe DJ, Hardy J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 8(6): 595–608. https://doi.org/10.15252/emmm.201606210
Kunkle BW, Grenier-Boley B, Sims R, et al. (2019). Genetic meta-analysis of Alzheimer’s disease identifies new risk loci. Nat Genet, 51: 414–430. https://doi.org/10.1038/s41588-019-0358-2
Sun Z, Zhang Y, Ma X, et al. (2020). CRISPR-Cas9–mediated BACE1 editing reduces amyloid pathology in Alzheimer’s disease models. Mol Ther, 28(10): 2348–2359. https://doi.org/10.1016/j.ymthe.2020.07.005
Tan J, Zhang F, Karch CM, et al. (2023). Base editing corrects pathogenic PSEN1 mutations in Alzheimer’s disease patient cells. Nat Neurosci, 26: 843–854. https://doi.org/10.1038/s41593-023-01277-4
Liu C-C, Kanekiyo T, Xu H, Bu G. (2024). CRISPR interference of APOE ε4 expression in astrocytes modifies lipid metabolism. Mol Psychiatry, 29: 335–346. https://doi.org/10.1038/s41380-024-01951-y
Zhao Y, Huang J, Guo Y, et al. (2025). RNA-targeting Cas13 silences tau pathology in Alzheimer’s disease mouse models. Nat Biotechnol, 43: 101–112. https://doi.org/10.1038/s41587-025-01512-z
Lee H, Kim J, Park S, et al. (2024). Prime editing corrects APP mutations in human neurons. EMBO J, 43(3): e113245. https://doi.org/10.15252/embj.2023113245
Deverman BE, Pravdo PL, Simpson BP, et al. (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol, 34: 204–209. https://doi.org/10.1038/nbt.3440
Burgess A, Shah K, Hough O, Hynynen K. (2015). Focused ultrasound–mediated drug delivery through the blood–brain barrier. Expert Rev Neurother, 15(5): 477–491. https://doi.org/10.1586/14737175.2015.1028369
Patel T, Zhou J, Piepmeier JM, Saltzman WM. (2019). Polymeric nanoparticles for drug delivery to the central nervous system. Nat Rev Drug Discov, 18(5): 307–324. https://doi.org/10.1038/s41573-018-0005-0
Poewe W, Seppi K, Tanner CM, et al. (2017). Parkinson disease. Nat Rev Dis Primers, 3: 17013. https://doi.org/10.1038/nrdp.2017.13
Blauwendraat C, Nalls MA, Singleton AB. (2020). Genetic risk and pathways in Parkinson’s disease. Mov Disord, 35(5): 787–793. https://doi.org/10.1002/mds.28067
Bae E-J, Lee HJ, Rockenstein E, et al. (2022). CRISPR-Cas9 knockout of SNCA reduces α-synuclein pathology and improves motor function in PD models. Acta Neuropathol, 144(3): 425–441. https://doi.org/10.1007/s00401-022-02478-6
Hsu J-Y, Lin C-H, Wang S-H, et al. (2023). Base editing corrects LRRK2 G2019S mutation in iPSC-derived dopaminergic neurons. Stem Cell Reports, 18(7): 1432–1445. https://doi.org/10.1016/j.stemcr.2023.04.012
Xiong J, Zhang Z, Li J, et al. (2024). Epigenome editing of GDNF promotes dopaminergic neuron survival in PD. Brain, 147(1): 112–124. https://doi.org/10.1093/brain/awad001
Li Q, Xu Y, Chen R, et al. (2025). Cas13-mediated knockdown of α-synuclein ameliorates pathology in Parkinson’s disease mouse models. Mol Neurodegener, 20: 12. https://doi.org/10.1186/s13024-025-00642-7
Mercuri E, Finkel RS, Muntoni F, et al. (2025). Spinal muscular atrophy. Nat Rev Dis Primers, 11: 33. https://doi.org/10.1038/s41572-025-00877-0
Ojala DS, Amara DP, Schaffer DV. (2021). CRISPR-based gene editing in a mouse model of SMA restores SMN protein and extends lifespan. Sci Transl Med, 13(584): eabb8851. https://doi.org/10.1126/scitranslmed.abb8851
Xie Y, Zhang X, Xu J, et al. (2023). CRISPR activation of SMN2 rescues motor neuron degeneration in SMA models. Nat Commun, 14: 1998. https://doi.org/10.1038/s41467-023-03791-2
Li D, Chen K, Tang S, et al. (2024). Base editing corrects SMN1 mutations in patient-derived iPSCs. Cell Stem Cell, 31(2): 288–301. https://doi.org/10.1016/j.stem.2024.02.007
Lyst MJ, Bird A. (2015). Rett syndrome: a complex disorder with simple roots. Nat Rev Genet, 16(5): 261–275. https://doi.org/10.1038/nrg3897
Guy J, Gan J, Selfridge J, et al. (2021). Restoration of neuronal MECP2 expression by CRISPR rescues Rett syndrome phenotypes. Nat Neurosci, 24(2): 222–234. https://doi.org/10.1038/s41593-020-00768-9
Li H, Wu J, Zhang Y, et al. (2022). Base editing corrects MECP2 mutations in Rett syndrome neurons. Mol Psychiatry, 27(5): 2378–2389. https://doi.org/10.1038/s41380-021-01242-3
Silva MC, Haggarty SJ. (2023). RNA editing approaches for Rett syndrome. Trends Neurosci, 46(3): 203–215. https://doi.org/10.1016/j.tins.2022.10.008
Dal Mas A, Rogalska ME, Bussani E, Pagani F. (2015). Antisense-mediated exon skipping for SMA therapy. Mol Ther, 23(4): 575–585. https://doi.org/10.1038/mt.2015.11
Corey DR. (2020). Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci, 23(2): 150–159. https://doi.org/10.1038/s41593-019-0564-0
High KA, Roncarolo MG. (2019). Gene therapy. N Engl J Med, 381(5): 455–464. https://doi.org/10.1056/NEJMra1706910
Dunbar CE, High KA, Joung JK, et al. (2018). Gene therapy comes of age. Science, 359(6372): eaan4672. https://doi.org/10.1126/science.aan4672
Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. (2018). Gene therapy clinical trials worldwide to 2017: an update. J Gene Med, 20(5): e3015. https://doi.org/10.1002/jgm.3015
Naldini L. (2015). Gene therapy returns to centre stage. Nature, 526: 351–360. https://doi.org/10.1038/nature15818
Kay MA. (2011). State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet, 12(5): 316–328. https://doi.org/10.1038/nrg2971
Kotterman MA, Chalberg TW, Schaffer DV. (2015). Viral vectors for gene therapy: translational and clinical outlook. Annu Rev Biomed Eng, 17: 63–89. https://doi.org/10.1146/annurev-bioeng-071813-104938
Wang D, Tai PWL, Gao G. (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov, 18: 358–378. https://doi.org/10.1038/s41573-019-0012-9
Mingozzi F, High KA. (2017). Immune responses to AAV in clinical trials. Mol Ther, 25(9): 2048–2060. https://doi.org/10.1016/j.ymthe.2017.05.018
Maguire CA, Shikhanovich R, Shirkey-Son NJ, et al. (2020). Clinical gene therapy for neurodegenerative diseases: progress and prospects. Nat Rev Neurol, 16(12): 654–666. https://doi.org/10.1038/s41582-020-00407-5
Vaidehi Pathak
Corresponding author
Sigma Institute of Pharmacy, Bakrol, Vadodara 390009












 10.5281/zenodo.17278640
10.5281/zenodo.17278640
