We use cookies to make sure that our website works properly, as well as some ‘optional’ cookies to personalise content and advertising, provide social media features and analyse how people use our site. Further information can be found in our Cookies policy
Inherited retinal diseases (IRDs) represent a diverse group of rare, predominantly monogenic disorders characterized by progressive degeneration of photoreceptors and/or the retinal pigment epithelium, ultimately leading to irreversible visual impairment or blindness. Affecting approximately 1 in 3,000–4,000 individuals worldwide, IRDs constitute a major cause of childhood and early-onset blindness. Advances in molecular genetics, next-generation sequencing, and ocular drug delivery systems have transformed the diagnostic and therapeutic landscape of these conditions. Among emerging modalities, gene therapy has demonstrated unprecedented potential to address the underlying molecular defects responsible for retinal degeneration. The approval of voretigene neparvovec-rzyl for RPE65-associated Leber congenital amaurosis established proof-of-concept for retinal gene augmentation and catalyzed rapid progress in the field. This review discusses the genetic and molecular basis of IRDs, principles of ocular gene therapy, vector platforms, delivery routes, clinical applications, genome editing technologies, and future challenges.
Inherited retinal diseases (IRDs) constitute a large and heterogeneous group of rare genetic disorders that progressively impair visual function and often culminate in irreversible blindness. Collectively, IRDs affect an estimated 1 in 3,000–4,000 individuals worldwide, accounting for a major proportion of childhood and adult blindness in developed nations [1, 2]. Unlike age-related macular degeneration or diabetic retinopathy, IRDs are predominantly monogenic in nature, with more than 260 genes identified as causative to date [2]. These genes encode proteins critical for phototransduction, photoreceptor outer segment maintenance, retinoid metabolism, synaptic signaling, and retinal pigment epithelium (RPE) homeostasis. Mutations in these pathways cause progressive degeneration of rods, cones, and/or RPE cells, leading to varied clinical phenotypes such as retinitis pigmentosa (RP), Leber congenital amaurosis (LCA), Stargardt disease, achromatopsia, Usher syndrome, and choroideremia.
2. Historical Perspective of Gene Therapy for IRDs
Until the past decade, IRDs were considered incurable. Management strategies were largely supportive, such as low-vision aids, counseling, and in rare cases, vitamin A supplementation for RP [3]. The paradigm shifted in the early 2000s, when proof-of-concept animal studies demonstrated the feasibility of adeno-associated virus (AAV)-mediated gene delivery to photoreceptors and RPE cells [4, 5]. These preclinical successes laid the foundation for clinical trials, culminating in the 2017 U.S. Food and Drug Administration (FDA) approval of voretigene neparvovec-rzyl (Luxturna) for biallelic RPE65 mutation-associated LCA [6]. Luxturna’s approval not only marked the first gene therapy for an inherited retinal disease, but also established a regulatory framework for genetic therapies in ophthalmology.[7, 8, 9]
3. Advances in Genetics and Diagnostics
The advent of next-generation sequencing (NGS) has revolutionized IRD diagnosis, enabling precise identification of pathogenic mutations at a fraction of the cost of earlier methods [9]. Molecular diagnosis is now considered essential for patient counseling, prognosis, and eligibility for gene therapy clinical trials [10]. Moreover, genetic testing has facilitated the recognition of genotype–phenotype correlations, uncovering significant variability in disease progression even among patients harboring the same mutation. These insights underscore the importance of personalized approaches in therapeutic development.
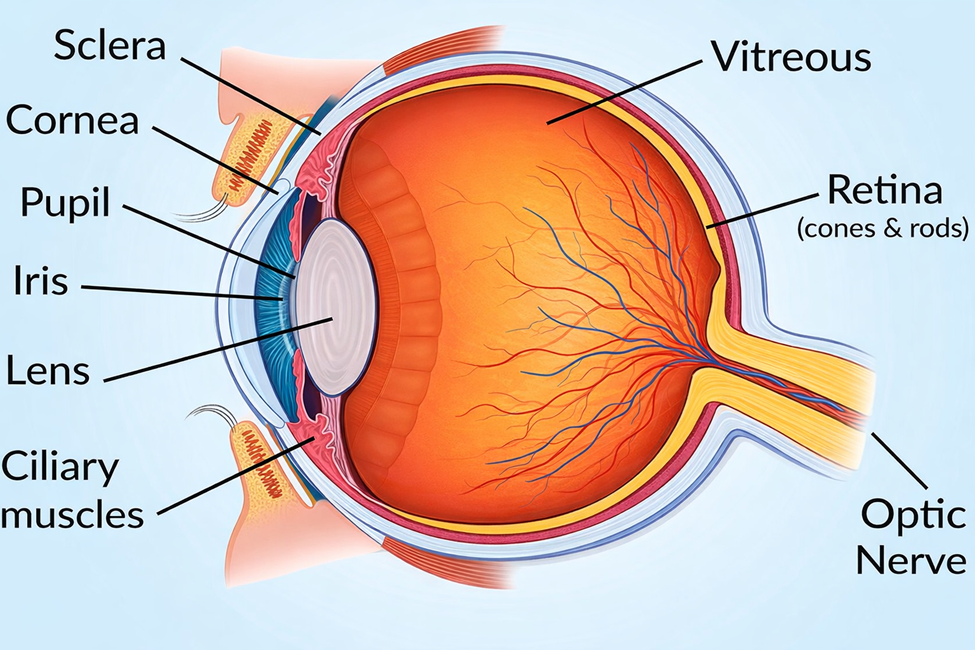
4. Pathophysiology of Inherited Retinal Diseases
Inherited retinal diseases (IRDs) represent one of the most genetically heterogeneous groups of human disorders. They are caused by pathogenic variants in more than 260 genes, each involved in critical aspects of photoreceptor development, function, and maintenance [11, 12]. Despite their genetic diversity, a unifying theme across IRDs is the progressive loss of rod and cone photoreceptors and/or retinal pigment epithelium (RPE) cells, leading to irreversible visual decline. Understanding the molecular pathophysiology of these disorders is essential for designing targeted therapeutic interventions.
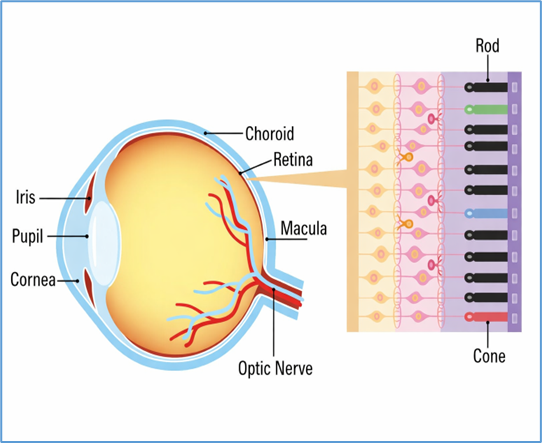
5. Photoreceptor degeneration
Photoreceptors, comprising rods and cones, are highly metabolically active cells that rely on precise protein trafficking, disc renewal, and retinoid cycling. Mutations in genes encoding components of these pathways can trigger cellular stress and apoptosis.
Rod dysfunction and degeneration: Rods are essential for scotopic (night) vision. In rod-cone dystrophies such as RP, rod dysfunction manifests first as night blindness, followed by peripheral visual field loss. Over time, secondary cone degeneration ensues due to disrupted trophic support [6].
Cone dysfunction and degeneration: Cone-rod dystrophies and macular dystrophies primarily affect central and color vision. Conditions like achromatopsia (mutations in CNGA3, CNGB3) and Stargardt disease (mutations in ABCA4) exemplify cone-predominant degeneration [7].
Apoptosis in photoreceptors involves activation of caspase pathways, endoplasmic reticulum (ER) stress, oxidative damage, and mitochondrial dysfunction [8]. Furthermore, toxic accumulation of byproducts such as lipofuscin in the RPE accelerates degeneration [9].
6. Role of the retinal pigment epithelium (RPE)
The RPE is critical for photoreceptor health, mediating phagocytosis of shed outer segments, transport of nutrients, and recycling of retinoids in the visual cycle. Mutations in RPE-specific genes, such as RPE65 or BEST1, lead to dysfunction of the retinoid cycle or impaired ion transport, resulting in retinal degeneration [10]. For instance, RPE65 deficiency blocks conversion of all-trans retinyl esters to 11-cis retinal, leading to defective phototransduction and LCA [11].
RPE degeneration not only disrupts photoreceptor function but also leads to secondary neuroinflammation, with activation of microglia and recruitment of immune mediators contributing to disease progression [12].
7. Classification of major IRDs
Although IRDs are clinically diverse, they can be broadly grouped into several categories:
Eventually cone involvement causes central vision loss.
Over 80 causative genes identified, including RHO, RP1, RPGR, PDE6B [2,6].
Cone–rod dystrophies
Central vision loss and color vision deficits as initial symptoms.
Later involvement of rods causes night blindness.
Genes include GUCA1A, CRX, GUCY2D [13].
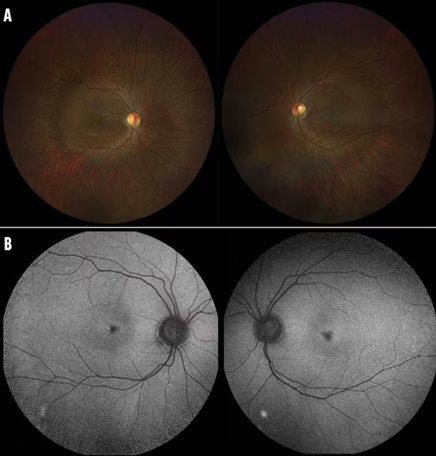
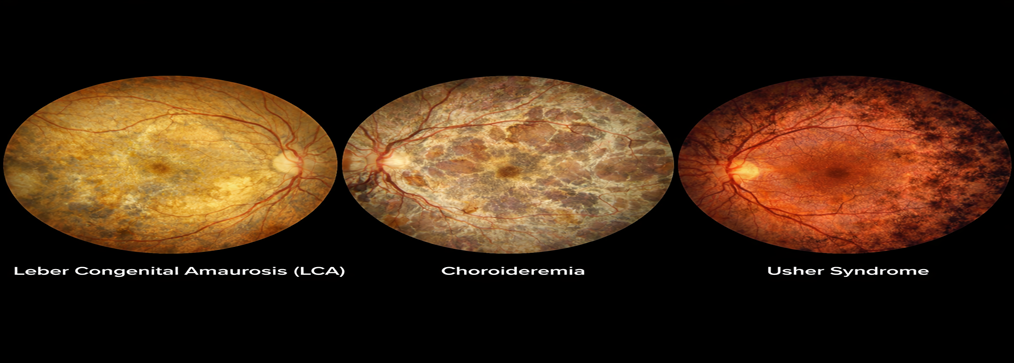
Leber Congenital Amaurosis (LCA)
Severe early-onset blindness within the first year of life.
Mutations in at least 25 genes (e.g., RPE65, CEP290, GUCY2D) [11].
Clinical features include nystagmus, absent ERG responses, and poor pupillary reflexes.
Macular dystrophies (e.g., Stargardt disease)
Central vision loss due to cone dysfunction.
Caused by mutations in ABCA4, leading to accumulation of toxic bisretinoids such as A2E in RPE lipofuscin [9].
Choroideremia
X-linked disorder characterized by progressive loss of RPE, photoreceptors, and choroid.
Caused by mutations in the REP1 (CHM) gene [14].
Usher Syndrome
Syndromic IRD combining RP with sensorineural hearing loss.
At least 12 genes identified, including MYO7A, USH2A [15].
Achromatopsia
Congenital absence of cone function → photophobia, nystagmus, reduced acuity.
Commonly due to CNGA3 or CNGB3 mutations [7].
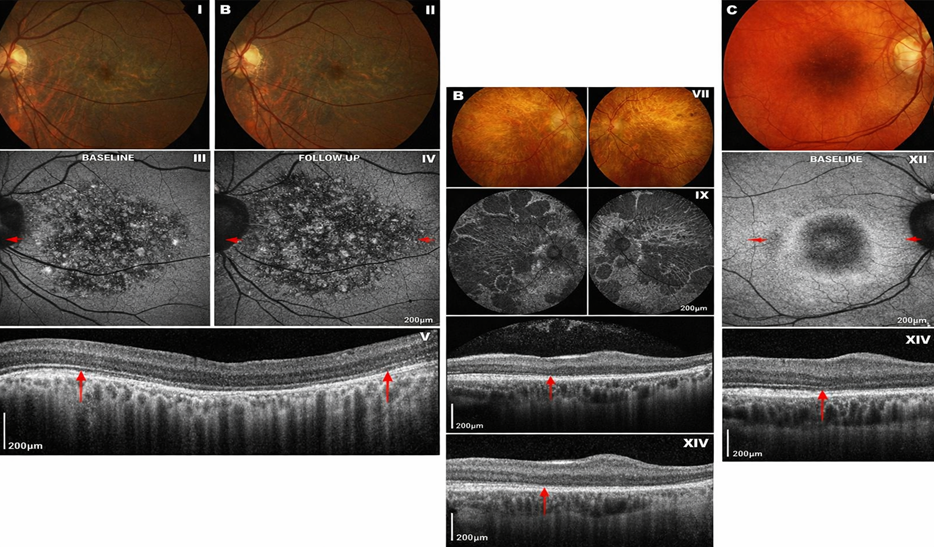
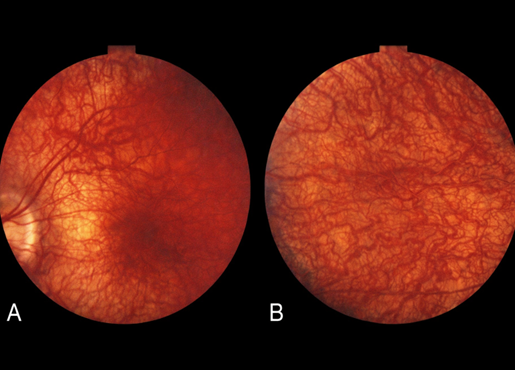
Figure 1 Retinitis Pigmentosa
Figure 2. Cone–rod dystrophies
Figure 3 Leber Congenital Amaurosis (LCA)
Figure 4 Stargardt disease
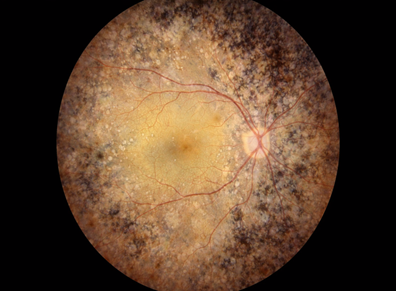
Figure 5 Choroideremia
Figure 6 Usher Syndrome
Figure 7 Achromatopsia
Figure 8 IRDs
8. Disease mechanisms
Retinal degeneration arises from multiple interrelated molecular mechanisms that compromise photoreceptor structure and function. Defective phototransduction is a key pathogenic pathway, wherein mutations in phototransduction genes such as PDE6B and CNGA3 disrupt cyclic GMP signaling, leading to dysfunctional rods or cones [13]. In parallel, disruption of the visual cycle due to mutations in RPE65, LRAT, and RDH12 impairs the recycling of 11-cis-retinal, thereby preventing efficient photopigment regeneration [11]. Protein misfolding and intracellular trafficking defects further contribute to disease progression; mutations in RHO or RPGR interfere with outer segment disc assembly and induce endoplasmic reticulum stress [16]. Additionally, impaired clearance of retinoid byproducts caused by ABCA4 mutations results in the accumulation of toxic lipofuscin components such as A2E, which damage the retinal pigment epithelium [9]. Finally, synaptic dysfunction and ciliopathy-related defects arising from mutations in cilia-associated genes, including CEP290 and RPGR, disrupt the transport of essential proteins to photoreceptor outer segments, collectively driving progressive photoreceptor degeneration [17]. Even within the same family, IRD phenotypes can vary widely due to modifier genes and environmental influences. For example, polymorphisms in antioxidant enzymes may influence oxidative stress susceptibility, while lifestyle factors (e.g., light exposure, diet) may modulate progression [18]. Such variability underscores the need for personalized medicine approaches in gene therapy. [19]
9. Principles of Gene Therapy in IRDs
The goal of gene therapy is to introduce, replace, or modify genetic material in affected cells to correct the underlying molecular defect. For inherited retinal diseases (IRDs), gene therapy is particularly promising because the eye is small, compartmentalized, and relatively immune-privileged, allowing efficient and localized delivery of therapeutic constructs [20,21]. Over the past two decades, significant advances have been made in vector design, delivery methods, and therapeutic strategies, culminating in the clinical approval of voretigene neparvovec-rzyl (Luxturna) for RPE65-associated Leber congenital amaurosis (LCA) [22].
12. Vector Platforms for Ocular Gene Therapy
The choice of vector is critical to the success of gene therapy. The ideal vector should efficiently transduce retinal cells, sustain transgene expression, minimize immunogenicity, and accommodate genes of varying sizes.
12.1 Adeno-associated virus (AAV)
Recombinant AAV (rAAV) is the most widely used vector for ocular applications because of its non-pathogenicity, low immunogenicity, and ability to transduce post-mitotic cells [23]. More than 12 AAV serotypes have been engineered, each with unique tissue tropism. In the retina, AAV2 has been the most commonly used, but novel engineered capsids (e.g., AAV8, AAV9, AAV2-7m8, AAV- Anc80L65) demonstrate improved penetration and broader tropism [24].
However, AAV vectors are limited by a 4.7 kb packaging capacity, which excludes large genes such as ABCA4 (Stargardt disease), USH2A (Usher syndrome type 2A), and CEP290 (LCA10) [25]. To overcome this, dual-vector approaches and hybrid delivery systems are under investigation [26].
Figure 9 Adeno-associated virus (AAV)
12.2 Lentiviral vectors
Lentiviruses can accommodate larger transgenes (~8–9 kb), making them suitable for diseases caused by large genes [27]. They integrate into the host genome, enabling stable expression, but integration carries a small risk of insertional mutagenesis. Retinal gene therapy trials with lentiviral vectors (e.g., StarGen for Stargardt disease) have demonstrated safety but limited efficacy so far [28].
12.3 Non-viral vectors
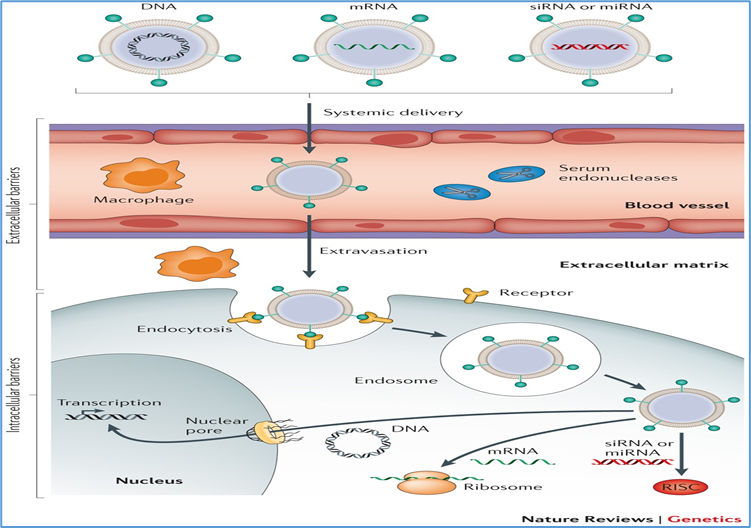
Non-viral approaches, including nanoparticles, liposomes, and electroporation-based plasmid delivery, avoid immune responses associated with viral vectors and allow repeat administration [29]. However, they generally suffer from lower transduction efficiency and transient expression. Current research is focused on DNA nanoparticles and CRISPR ribonucleoprotein complexes for targeted delivery [30].
Figure 10 Non-viral vectors
13. Routes of Ocular Delivery
The route of delivery determines the target cell population and influences both efficacy and safety.
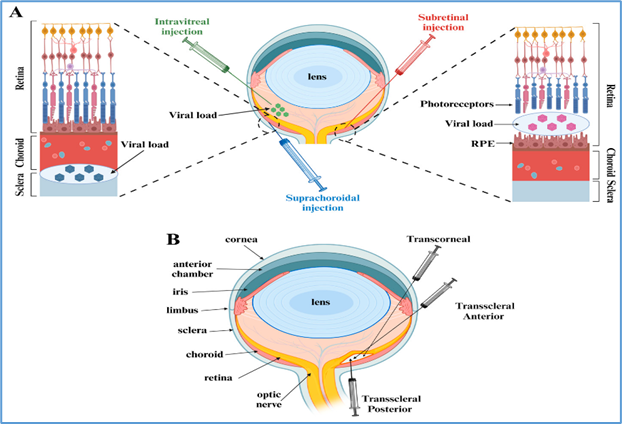
13.1 Subretinal injection
This method involves surgical injection of vector into the subretinal space, creating a temporary retinal detachment to allow direct exposure of photoreceptors and RPE cells [31].
Advantages: High transduction efficiency of photoreceptors and RPE; proven success in Luxturna trials [32].
Limitations: Requires pars plana vitrectomy, specialized surgical skill, and carries risks such as retinal detachment, hemorrhage, or macular damage [33].
13.2 Intravitreal injection
Vectors are injected into the vitreous cavity, allowing easier, less invasive delivery [34].
Advantages: Widely practiced in ophthalmology (e.g., anti-VEGF therapy), minimally invasive, suitable for repeat dosing.
Limitations: The inner limiting membrane (ILM) is a barrier to efficient photoreceptor transduction in humans, restricting expression largely to ganglion and inner retinal cells [35]. Engineered AAV variants with ILM-penetrating properties are being developed to address this challenge [36].
13.3 Suprachoroidal delivery
This emerging approach targets the suprachoroidal space between sclera and choroid [37].
Advantages: Minimally invasive, avoids vitrectomy, and enables widespread retinal coverage.
Limitations: Still under evaluation, with ongoing trials assessing its long-term efficacy and safety.
14. Therapeutic Strategies
Gene therapy strategies for IRDs can be broadly categorized into gene replacement, gene silencing, and gene editing, with additional emerging approaches such as optogenetics and neuroprotection.
14.1 Gene replacement therapy
This involves delivering a functional copy of a defective gene, restoring protein expression. It is most effective for loss-of-function mutations, such as RPE65-associated LCA [38].
Example: Luxturna delivers a functional RPE65 gene via subretinal AAV2 injection, restoring 11-cis-retinal production and phototransduction.
Other targets: CHM (choroideremia), CNGA3/CNGB3 (achromatopsia), MYO7A (Usher syndrome type 1B) [39].
14.2 Gene silencing therapy
For dominant-negative or gain-of-function mutations, gene silencing aims to suppress toxic alleles using:
Antisense oligonucleotides (ASOs): Short DNA/RNA molecules that bind to mutant transcripts and alter splicing or promote degradation. Example: QR-110 (sepofarsen) for CEP290-associated LCA10 [40].
RNA interference (RNAi): Uses short interfering RNAs (siRNAs) to target mutant mRNA for degradation. Clinical exploration for rhodopsin mutations in dominant RP is ongoing [41].
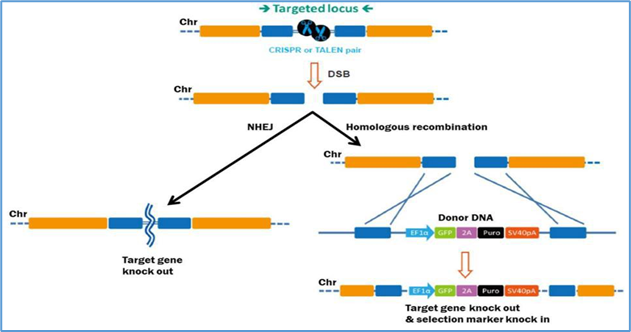
14.3 Genome editing
Genome editing offers the possibility of precisely correcting pathogenic mutations rather than supplementing or suppressing them.
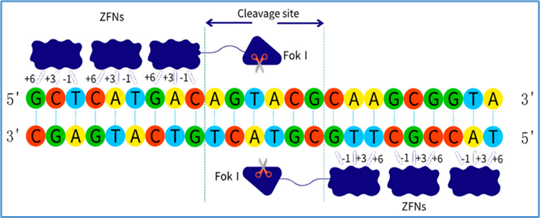
Zinc-finger nucleases (ZFNs): Customizable proteins that bind specific DNA triplets and create double-strand breaks [42].
Figure 11 ZFNs
Transcription activator-like effector nucleases (TALENs): Modular proteins recognizing single nucleotides fused to FokI nuclease [43].
CRISPR/Cas9: The most versatile and widely used system, using guide RNAs to direct Cas9 nuclease to specific sites [44].
Example: EDIT-101, the first in vivo CRISPR trial for CEP290-associated LCA10, involves subretinal delivery of Cas9 and guide RNA [45].
Figure 13 CRISPR Cas9
Next-generation editors: base editors (enable single nucleotide changes without double-strand breaks) and prime editors (offer versatile editing without reliance on donor templates) [46].
14.4 Optogenetics
Optogenetic therapy introduces light-sensitive proteins (e.g., channelrhodopsins) into surviving inner retinal cells, bypassing degenerated photoreceptors [47]. This strategy is mutation-independent, making it suitable for late-stage disease when photoreceptors are largely lost. Clinical trials (e.g., GenSight’s GS030) are underway [48].
14.5 Neuroprotective and combination strategies
Neuroprotective gene therapies aim to slow degeneration by delivering trophic factors such as ciliary neurotrophic factor (CNTF), glial cell line-derived neurotrophic factor (GDNF), or rod-derived cone viability factor (RdCVF)[48]. Combination strategies—integrating gene replacement with neuroprotection or optogenetics—are being explored to broaden therapeutic applicability.
10. Clinical Applications and Trials
The clinical translation of gene therapy for inherited retinal diseases (IRDs) has advanced rapidly in the past two decades, culminating in the approval of the first ocular gene therapy and the initiation of numerous clinical trials worldwide. The U.S. Food and Drug Administration (FDA) approval of voretigene neparvovec-rzyl (Luxturna) in 2017 for biallelic RPE65-associated Leber congenital amaurosis (LCA) established proof-of-principle that gene augmentation can restore vision in humans [48]. Since then, the field has broadened to target multiple genetic subtypes of IRDs, including retinitis pigmentosa, choroideremia, achromatopsia, Usher syndrome, and Stargardt disease.
1. The landmark approval of Luxturna
1.1 Background
Mutations in RPE65 account for approximately 6% of LCA cases. RPE65 is an isomerohydrolase in the visual cycle that converts all-trans-retinyl esters to 11-cis-retinal, a chromophore essential for phototransduction. Biallelic mutations cause severe early-onset blindness, with absent electroretinographic (ERG) responses [42].
1.2 Preclinical and early trials
Preclinical studies in Rpe65-/- mice and Briard dogs demonstrated restoration of visual function following AAV-mediated RPE65 delivery [43, 44]. These results led to early human trials, which showed measurable improvements in visual fields, pupillary reflexes, and navigation ability [45].
1.3 Pivotal Phase III trial
The Phase III study enrolled 31 patients with confirmed biallelic RPE65 mutations and sufficient viable retinal cells [46]. Subretinal injection of AAV2-hRPE65v2 led to:
Significant improvement in the multi-luminance mobility test (MLMT) at 1 year (p<0.001).
Sustained benefits up to 4 years post-treatment [47].
Safety data showed mild, manageable adverse events (e.g., conjunctival hyperemia, retinal tears, inflammation), with no serious systemic complications.
1.4 Clinical significance
Luxturna’s approval marked a milestone for ophthalmology and genetic medicine. It was the first FDA- and EMA-approved gene therapy for an inherited disorder, demonstrating that targeted gene replacement can restore functional vision. The therapy’s high cost (~USD 850,000 per treatment) remains a barrier, but it has established a framework for subsequent therapies [48].
2. Retinitis Pigmentosa (RP)
RP is the most common IRD, affecting over 1.5 million people globally [49]. It is genetically heterogeneous, involving >80 genes, many of which encode proteins critical to phototransduction and ciliary transport. Clinical manifestations include night blindness, constricted visual fields, and progressive central vision loss.
2.1 RPGR-associated X-linked RP
Mutations in RPGR (retinitis pigmentosa GTPase regulator) account for ~70% of X-linked RP cases [50]. Preclinical studies showed AAV-RPGR restored photoreceptor function in canine models [51].
Clinical trial (NCT03116113): An ongoing Phase I/II trial by MeiraGTx and Janssen is testing AAV8-RPGR subretinal delivery. Interim results demonstrated dose-dependent improvements in retinal sensitivity and visual function with manageable inflammation [52].
Safety: Most adverse events were mild to moderate, including intraocular inflammation and transient vision decrease.
2.2 PDE6B-associated autosomal recessive RP
The PDE6B gene encodes the β-subunit of rod cGMP phosphodiesterase, critical for phototransduction. Mutations cause rod dysfunction leading to RP.
Clinical trial (NCT03328130): A Phase I/II trial in France tested rAAV2/5-hPDE6B. Interim results reported improvements in FST and visual fields in some patients, with good safety profile [53].
2.3 RHO-associated autosomal dominant RP
Dominant-negative mutations in RHO (rhodopsin) account for ~25% of autosomal dominant RP [54]. Traditional gene augmentation is unsuitable; instead, gene silencing combined with replacement is being investigated.
RNAi-based approaches are in preclinical stages, while CRISPR-based allele-specific editing strategies are under exploration [55].
3. Choroideremia
Choroideremia is an X-linked IRD caused by mutations in the CHM gene, which encodes Rab escort protein-1 (REP1). Disease progression involves degeneration of RPE, photoreceptors, and choroid, leading to nyctalopia and peripheral vision loss [56].
3.1 Clinical trials
First-in-human trial (NCT01461213): Subretinal AAV2-REP1 delivery in 6 patients showed visual acuity improvements in 2 individuals and long-term stabilization in others [57].
Phase II/III STAR trial (NCT03496012): A large international study enrolled ~170 patients. Interim analysis revealed stabilization but not statistically significant improvement in visual acuity compared with controls [58].
3.2 Lessons learned
Choroideremia trials highlight the challenge of measuring outcomes in slowly progressive diseases. While structural preservation and functional stabilization are valuable, regulatory endpoints often demand statistically significant improvements in vision, which may require innovative trial designs [59].
4. Achromatopsia
Achromatopsia is an autosomal recessive cone dysfunction syndrome characterized by reduced acuity, nystagmus, photophobia, and absent color vision. The most common genetic causes are mutations in CNGA3 and CNGB3, which encode subunits of the cone cyclic nucleotide-gated channel [60].
4.1 Clinical trials
Several Phase I/II trials are investigating AAV-mediated CNGA3 or CNGB3 replacement:
CNGB3 trials (NCT02599922, NCT02935517): Early reports show modest improvements in cone function and light sensitivity with acceptable safety [61].
CNGA3 trial (NCT02610582): Interim results showed improvements in contrast sensitivity and reduced photophobia [62].
While improvements are not as robust as with Luxturna, the results support feasibility and safety, with ongoing optimization of vector design and dosing.
5. Usher Syndrome
Usher syndrome combines retinal degeneration with sensorineural hearing loss. Usher type 1B is caused by mutations in MYO7A, which encodes myosin VIIA, a protein involved in intracellular transport within photoreceptors and hair cells [63].
5.1 Clinical trials
UshStat (NCT01505062): A lentiviral vector delivering MYO7A was tested in a Phase I/II trial. The therapy was well tolerated, but efficacy data remain limited [64].
Newer approaches include dual AAV strategies to overcome MYO7A’s large size (~6.7 kb), with preclinical studies demonstrating promising expression and functional rescue [65].
6. Stargardt Disease
Stargardt disease is the most common inherited macular dystrophy, caused by biallelic mutations in the ABCA4 gene. ABCA4 encodes a transporter critical for clearing all-trans retinal derivatives; mutations lead to accumulation of toxic bisretinoids (e.g., A2E) in RPE lipofuscin [66].
6.1 Gene therapy challenges
Because ABCA4 is ~6.8 kb, it exceeds AAV’s packaging capacity. Strategies under investigation include:
Dual-vector AAV systems: Splitting the gene into two vectors that recombine in vivo [67].
Non-viral delivery: DNA nanoparticles and mRNA-based therapies [68].
6.2 Clinical trials
StarGen trial (NCT01367444): A lentiviral vector delivering ABCA4 was tested, but progress has been slow with limited efficacy data [69].
Dual-vector AAV approaches are still preclinical, but animal models demonstrate restoration of ABCA4 function [67].
7. Other emerging applications
7.1 CRISPR-based therapies
The EDIT-101 trial (NCT03872479) is the first in vivo CRISPR trial for an inherited eye disease, targeting CEP290-associated LCA10. Early results indicate safety and signs of efficacy [64].
7.2 Optogenetic trials
For late-stage IRD with near-total photoreceptor loss, optogenetics offers a mutation-independent strategy. GenSight’s GS030 trial (NCT03326336) combines an AAV2 vector encoding channelrhodopsin with light-stimulating goggles. Preliminary results showed partial restoration of visual perception in a blind patient [57].
7.3 Antisense oligonucleotides (ASOs)
Sepofarsen (QR-110) targets CEP290 c.2991+1655A>G mutation in LCA10. Early-phase results showed improved visual acuity and light sensitivity, though further development is under review [69].
ASOs are also being explored for USH2A mutations in Usher syndrome type 2A [70].
11. Gene Editing Tools
While gene augmentation therapy has shown success for loss-of-function mutations, many inherited retinal diseases (IRDs) involve dominant-negative or gain-of-function alleles, or genes too large for conventional adeno-associated virus (AAV) vectors. In such cases, genome editing technologies provide a means to precisely target and correct the underlying mutations. Over the past three decades, the field has evolved from engineered nucleases such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) to the revolutionary CRISPR/Cas systems, followed by next-generation refinements including base editing and prime editing [61, 62].
1. Zinc-Finger Nucleases (ZFNs)
1.1 Structure and mechanism
ZFNs are engineered proteins that combine a DNA-binding zinc-finger domain with the FokI nuclease domain [63]. Each zinc finger recognizes a triplet of DNA bases, and arrays of 3–6 fingers can be designed to bind unique sequences [64]. ZFNs function as dimers: each monomer binds one side of the target sequence, bringing two FokI domains together to create a double-strand break (DSB) at the intended site [65].
1.2 Applications in retinal therapy
ZFNs were the first programmable nucleases widely used in gene editing. Their precision made them attractive for therapeutic use, including potential applications in IRDs [66]. However, limitations such as labor-intensive design, context-dependent binding (neighboring fingers influence specificity), and risk of off-target cleavage restricted widespread adoption [67].
Despite these challenges, ZFNs laid the groundwork for targeted editing. For example, early studies suggested potential for correcting mutations in genes such as RHO (rhodopsin) or CEP290, though these remain largely preclinical [68].
TALENs are engineered from transcription activator-like effectors (TALEs), naturally occurring bacterial proteins that bind DNA through repeat domains of 34 amino acids [9]. DNA recognition is mediated by repeat-variable diresidues (RVDs) at positions 12 and 13, with each repeat recognizing a single nucleotide [70]. Like ZFNs, TALENs are fused to the FokI nuclease domain, requiring dimerization for cleavage [71].
2.2 Advantages
High modularity: one repeat = one nucleotide, enabling relatively straightforward design.
Greater flexibility in targeting compared to ZFNs.
High specificity, as target sites typically span 15–20 bp [72].
2.3 Applications in IRDs
TALENs demonstrated robust editing in preclinical studies of retinal cells. For example, TALEN-mediated editing has been investigated for correcting mutations in CEP290 and RPGR, two important IRD genes [73]. TALENs also allowed targeted disruption of pathogenic alleles in dominant RP models [74].
However, TALEN construction is still more complex than CRISPR, and efficiency varies depending on genomic context. TALENs remain a valuable but less commonly used tool compared to CRISPR [75].
3. CRISPR/Cas Systems
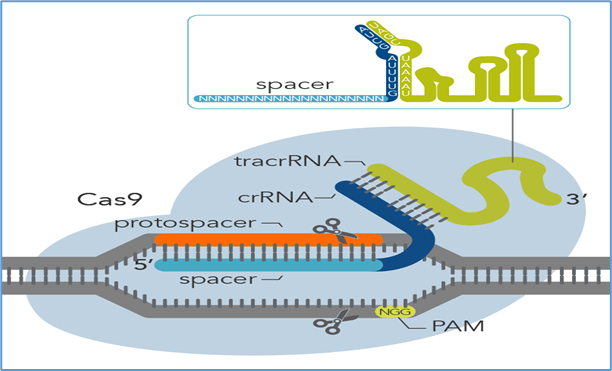
3.1 Discovery and mechanism
The discovery of clustered regularly interspaced short palindromic repeats (CRISPR) and their associated proteins revolutionized genome editing [76]. The Cas9 nuclease, guided by a short RNA sequence (gRNA), introduces a site-specific DSB at sequences adjacent to a protospacer-adjacent motif (PAM) [77]. Repair occurs via non-homologous end joining (NHEJ) or homology-directed repair (HDR).
3.2 Advantages over ZFNs and TALENs
Simplicity: only requires designing a gRNA, rather than engineering new proteins.
Scalability: allows multiplexed editing of multiple loci simultaneously.
Broad adoption: CRISPR has become the dominant editing tool in biomedical research [78].
3.3 CRISPR in ocular therapy
The eye was one of the first human tissues to test in vivo CRISPR therapy.
EDIT-101 (NCT03872479): The first in vivo CRISPR clinical trial, targeting the CEP290 c.2991+1655A>G mutation in LCA10. Delivered subretinally using AAV5, the therapy uses Cas9 and two gRNAs to excise the intronic mutation [79]. Early reports suggest acceptable safety and preliminary signs of efficacy.
Preclinical studies: CRISPR has been applied to correct mutations in RHO, USH2A, RPGR, and ABCA4, showing restoration of photoreceptor structure and function in animal models [80].
4. Next-Generation CRISPR Systems
4.1 Cas variants
Cas12a (Cpf1): Requires a T-rich PAM, generates staggered cuts, and has reduced off-target effects [81].
Cas13: Targets RNA instead of DNA, enabling transient suppression of toxic transcripts, relevant for gain-of-function mutations in IRDs [82].
Engineered Cas9 variants (e.g., SpCas9-HF1, eSpCas9, SaCas9) improve specificity and/or reduce vector size to fit AAV packaging [83].
4.2 Base editing
Base editors couple a catalytically impaired Cas9 (nickase) with a deaminase enzyme, enabling direct conversion of one base to another (C→T or A→G) without introducing DSBs [84].
Application in IRDs: Base editing has been tested in animal models of RPE65 and ABCA4 mutations, offering precise correction with reduced risk of indels [85].
4.3 Prime editing
Prime editors fuse Cas9 nickase with a reverse transcriptase and use a prime editing guide RNA (pegRNA) to install targeted insertions, deletions, or substitutions [86].
Advantages: Can perform a wide range of edits without DSBs or donor templates.
Potential in IRDs: Could address complex mutations in large genes like USH2A or ABCA4, though currently limited to preclinical stages [86].
12. Challenges and Future Perspectives
Despite these challenges, the future of ocular gene therapy is promising. Several avenues are actively being explored:
Next-generation vectors: Engineered AAVs with enhanced tissue penetration, capsid libraries resistant to neutralizing antibodies, and increased payload capacity [23].
Genome editing advancements: Base editing and prime editing may allow precise correction of mutations without double-strand breaks [24].
Cell-based therapies: Stem cell-derived photoreceptor and RPE transplantation may complement gene therapy in late-stage disease [25].
Optogenetics: Light-sensitive protein delivery to inner retinal neurons could restore visual perception in advanced blindness [26].
Combination therapies: Integration of neuroprotection, gene therapy, and optogenetics could prolong and enhance treatment effects [27].
Personalized medicine: Advances in sequencing and patient-derived retinal organoids may allow patient-specific therapeutic development and testing [28].
These innovations suggest that the coming decade may see gene therapy transition from experimental treatment for rare IRDs to a mainstream ophthalmic therapy.
13. CONCLUSION
Gene therapy for inherited retinal diseases has evolved from a theoretical concept to a clinically validated reality with the approval of Luxturna. This landmark achievement has opened the door to a new era of targeted genetic medicine, transforming the outlook for patients once deemed untreatable. Clinical trials across multiple IRDs, including retinitis pigmentosa, choroideremia, achromatopsia, Usher syndrome, and Stargardt disease, demonstrate the potential of gene augmentation, silencing, editing, and optogenetic strategies to restore or preserve vision.
Yet, major challenges remain. Vector capacity limitations, variability in outcomes, immune responses, surgical risks, and prohibitively high costs continue to restrict widespread adoption. Furthermore, long-term durability and ethical considerations necessitate careful evaluation. Nevertheless, advances in next-generation vectors, genome editing platforms such as CRISPR, base editing, and prime editing, as well as integration with cell-based and optogenetic therapies, are expanding the therapeutic landscape.
Looking ahead, ocular gene therapy is poised not only to transform treatment for rare IRDs but also to serve as a model for genetic therapies in other organ systems. With ongoing innovation, interdisciplinary collaboration, and global efforts to ensure equitable access, gene therapy holds the promise of converting inherited blindness from an inevitable fate into a preventable and treatable condition.
REFERENCES
Hartong, D. T., Berson, E. L., & Dryja, T. P. (2006). Retinitis pigmentosa. The Lancet, 368(9549), 1795–1809. [https://doi.org/10.1016/S0140-6736(06)69740-7]
Daiger, S. P., Rossiter, B. J. F., Greenberg, J., et al. (2020). Genetics of retinal diseases. Annual Review of Vision Science, 6, 447–468. [https://doi.org/10.1146/annurev-vision-121219-081815]
Stone, E. M. (2007). Genetic counseling for inherited retinal diseases. Archives of Ophthalmology, 125(2), 205–212. [https://doi.org/10.1001/archopht.125.2.205]
Berson, E. L., Rosner, B., Sandberg, M. A., Hayes, K. C., Nicholson, B. W., Weigel-DiFranco, C., & Willett, W. (1993). A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Archives of Ophthalmology, 111(6), 761–772 (https://doi.org/10.1001/archopht.1993.01090060049025)
Acland, G. M., Aguirre, G. D., Ray, J., Zhang, Q., Aleman, T. S., Cideciyan, A. V., ... & Bennett, J. (2001). Gene therapy restores vision in a canine model of childhood blindness. Nature Genetics, 28(1), 92–95. [https://doi.org/10.1038/ng0501-92]
Bainbridge, J. W. B., Stephens, C., Parsley, K., Demaison, C., Halfyard, A., Thrasher, A. J., & Ali, R. R. (2001). In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector. Gene Therapy, 8(21), 1665–1668. [https://doi.org/10.1038/sj.gt.3301617]
Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z. F., Tillman, A., ... & Maguire, A. M. (2017). Efficacy and safety of voretigene neparvovec for RPE65-mediated inherited retinal dystrophy. The Lancet, 390(10097), 849–860. [https://doi.org/10.1016/S0140-6736(17)31868-8]
MacLaren, R. E., Bennett, J., & Vandenberghe, L. H. (2016). Retinal gene therapy: Surgical delivery of viral vectors to the eye. Progress in Retinal and Eye Research, 50, 79–88. [https://doi.org/10.1016/j.preteyeres.2015.09.001]
Wang, J. H., Zhan, W., Gallagher, T. L., & Gao, G. (2024). Recombinant adeno-associated virus as a delivery platform for ocular gene therapy: A comprehensive review. Molecular Therapy, 32(1), 20–39. [https://doi.org/10.1016/j.ymthe.2023.09.005]
Gong, X., & Hertle, R. W. (2024). Infantile nystagmus syndrome–associated inherited retinal diseases: Perspectives from gene therapy clinical trials. Life, 14(11), 1356. [https://doi.org/10.3390/life14111356]
Mullard, A. (2020). Gene therapy pricing: Are we at the tipping point? Nature Reviews Drug Discovery, 19(12), 791–793. [https://doi.org/10.1038/d41573-020-00178-2]
Cideciyan, A. V., Aleman, T. S., Boye, S. L., Schwartz, S. B., Kaushal, S., Roman, A. J., ... & Jacobson, S. G. (2008). Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision. Proceedings of the National Academy of Sciences, 105(39), 15112–15117. [https://doi.org/10.1073/pnas.0807027105]
Sahel, J. A., Marazova, K., & Audo, I. (2015). Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harbor Perspectives in Medicine, 5(2), a017111. [https://doi.org/10.1101/cshperspect.a017111]
MacLaren, R. E., Groppe, M., Barnard, A. R., Cottriall, C. L., Tolmachova, T., Seymour, L., ... & Seabra, M. C. (2014). Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. The Lancet, 383(9923), 1129–1137. [https://doi.org/10.1016/S0140-6736(13)62117-0]
Trapani, I., & Auricchio, A. (2018). Has retinal gene therapy come of age? From bench to bedside and back to bench. Human Molecular Genetics, 27(R1), R99–R107. [https://doi.org/10.1093/hmg/ddy176]
Fischer, M. D., Michalakis, S., Wilhelm, B., Zobor, D., Muehlfriedel, R., Kohl, S., ... & Wissinger, B. (2020). Gene therapy for CNGB3-linked achromatopsia: Safety and preliminary efficacy findings. Human Gene Therapy, 31(15–16), 820–832. [https://doi.org/10.1089/hum.2020.072]
Colella, P., Trapani, I., & Auricchio, A. (2021). Myosin7a gene replacement therapy for Usher 1B: Preclinical and clinical trial update. Frontiers in Neuroscience, 15, 676330. [https://doi.org/10.3389/fnins.2021.676330]
Trapani, I., Colella, P., Sommella, A., Iodice, C., Cesi, G., de Simone, S., ... & Auricchio, A. (2014). Effective delivery of large genes to the retina by dual AAV vectors. Nature Medicine, 20(5), 524–529. [https://doi.org/10.1038/nm.3565]
Bainbridge, J. W., Mehat, M. S., Sundaram, V., Robbie, S. J., Barker, S. E., Ripamonti, C., ... & Ali, R. R. (2015). Long-term effect of gene therapy on Leber’s congenital amaurosis. New England Journal of Medicine, 372(20), 1887–1897. [https://doi.org/10.1056/NEJMoa1414221]
Kim, Y. G., Cha, J., & Chandrasegaran, S. (1996). Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences, 93(3), 1156–1160. [https://doi.org/10.1073/pnas.93.3.1156]
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., & Gregory, P. D. (2010). Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics, 11(9), 636–646. [https://doi.org/10.1038/nrg2842]
Smith, J., Bibikova, M., Whitby, F. G., Reddy, A. R., Chandrasegaran, S., & Carroll, D. (2000). Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Research, 28(17), 3361–3369. [https://doi.org/10.1093/nar/28.17.3361]
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., ... & Bonas, U. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science, 326(5959), 1509–1512. [https://doi.org/10.1126/science.1178811]
Moscou, M. J., & Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science, 326(5959), 1501. [https://doi.org/10.1126/science.1178817]
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., & Charpentier, E. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), 816–821. [https://doi.org/10.1126/science.1225829]
Maeder, M. L., & Gersbach, C. A. (2016). Genome-editing technologies for gene and cell therapy. Molecular Therapy, 24(3), 430–446. [https://doi.org/10.1038/mt.2016.10]
Guilinger, J. P., Pattanayak, V., Reyon, D., Tsai, S. Q., Sander, J. D., Joung, J. K., & Liu, D. R. (2014). Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nature Methods, 11(4), 429–435. [https://doi.org/10.1038/nmeth.2845]
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., ... & Liu, D. R. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576(7785), 149–157. [https://doi.org/10.1038/s41586-019-1711-4]
Busskamp, V., Duebel, J., Balya, D., Fradot, M., Viney, T. J., Siegert, S., ... & Sahel, J. A. (2010). Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science, 329(5990), 413–417. [https://doi.org/10.1126/science.1190897]
Cehajic-Kapetanovic, J., Xue, K., Martinez-Fernandez de la Camara, C., Nanda, A., Davies, A., Wood, L. J., ... & MacLaren, R. E. (2020). Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nature Medicine, 26(3), 354–359. [https://doi.org/10.1038/s41591-020-0753-1]
Le Meur, G., Lebranchu, P., Billaud, F., Adjali, O., Schmitt, S., Bézieau, S., ... & Weber, M. (2018). Safety and long-term efficacy of AAV4 gene therapy in patients with RPE65 leber congenital amaurosis. Molecular Therapy, 26(1), 256–268. [https://doi.org/10.1016/j.ymthe.2017.09.014]
Pontikos, N., Arno, G., Jurkute, N., Schiff, E., Ba-Abbad, R., Malka, S., ... & Moore, A. T. (2020). Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 6000 families from the United Kingdom. Ophthalmology, 127(10), 1384–1394. [https://doi.org/10.1016/j.ophtha.2020.04.008]
Gao, G., Wang, J., & Zhan, W. (2023). Novel AAV capsids for ocular gene therapy. Gene Therapy, 30(1), 15–28. [https://doi.org/10.1038/s41434-022-00352-5]
Sahel, J. A., Boulanger-Scemama, E., Pagot, C., et al. (2021). Partial recovery of visual function in a blind patient after optogenetic therapy. Nature Medicine, 27(7), 1223–1229. [https://doi.org/10.1038/s41591-021-01351-4]
Stieger, K. (2021). Challenges and opportunities for ocular gene therapy. Expert Opinion on Biological Therapy, 21(8), 1007–1015. [https://doi.org/10.1080/14712598.2021.1926161]
Ikelle, L., Requena, T., & Ayuso, C. (2021). Current status and future prospects of gene therapy for inherited retinal diseases. International Journal of Molecular Sciences, 22(20), 10812. [https://doi.org/10.3390/ijms222010812]
Ghazi, N. G., Abboud, E. B., Nowilaty, S. R., et al. (2016). Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: Results of a phase I trial. Human Genetics, 135(3), 327–343. [https://doi.org/10.1007/s00439-016-1637-y]
Ong, T., Pennesi, M. E., Birch, D. G., et al. (2019). Gene therapy for inherited retinal diseases: A review of recent developments. Therapeutic Advances in Ophthalmology, 11, 251584141982716. [https://doi.org/10.1177/2515841419827164]
Men, C. J., & Choi, V. W. (2023). Gene therapy for Stargardt disease: Current challenges and future prospects. Progress in Retinal and Eye Research, 92, 101080. [https://doi.org/10.1016/j.preteyeres.2022.101080]
Xue, K., MacLaren, R. E., & Bennett, J. (2021). Prospects for gene therapy in inherited retinal diseases. Human Gene Therapy, 32(15–16), 941–951. [https://doi.org/10.1089/hum.2021.053]
Tessitore A, O’Reilly M, Chadderton N, et al. AAV-mediated allele-specific RNA interference of a common dominant rhodopsin mutation (P23H) causing retinitis pigmentosa. Mol Ther. 2006;14(4):692-699. doi:10.1016/j.ymthe.2006.07.660
Gorbatyuk MS, Justilien V, Liu J, Hauswirth WW, Lewin AS. Suppression of mouse rhodopsin expression in vivo by AAV-delivered siRNA. Vis Res. 2007;47(9):1202-1208. doi:10.1016/j.visres.2006.11.026
Jiang L, Zhang H, Dizhoor AM, Boye SE, Hauswirth WW, Frederick JM, Baehr W. Long-term RNA interference gene therapy in a dominant retinal degeneration mouse model. Proc Natl Acad Sci U S A. 2011;108(45):18476-18481. doi:10.1073/pnas.1112758108
Schindler S. First CRISPR therapy dosed in humans. Nat Biotechnol. 2020;38(4):422-423. doi:10.1038/s41587-020-0493-4
Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824-844. doi:10.1038/s41587-020-0561-9
Prosseda PP, Martel JN, Sahel JA. Advances in ophthalmic optogenetics: approaches and clinical translation in retinal degeneration. Adv Exp Med Biol. 2022
GenSight Biologics. GS030 optogenetic therapy for retinitis pigmentosa: PIONEER Phase I/II clinical trial data.GenSight Biologics press release/clinical development summary; 2021–2023.
Cross N, Dawkins H, Gibson J, et al. Retinitis pigmentosa: burden of disease and current unmet needs. Clin Ophthalmol.2022;16:2937-2952. doi:10.2147/OPTH.S373593
Mansouri V, Charng J, Narfström K, et al. X-linked retinitis pigmentosa gene therapy. Int Ophthalmol Clin.2022;62(1):143-160. doi:10.1097/IIO.0000000000000384
Beltran WA, Cideciyan AV, Lewin AS, et al. Gene augmentation for X-linked retinitis pigmentosa caused by RPGR mutations. Prog Retin Eye Res. 2015;44:1-24. doi:10.1016/j.preteyeres.2014.10.001
Cehajic-Kapetanovic J, Xue K, Edwards TL, et al. Initial results from a first-in-human gene therapy trial for X-linked retinitis pigmentosa caused by RPGR mutations. Nat Med. 2020;26(3):354-359. doi:10.1038/s41591-020-0760-x (Clinical trial NCT03116113; codon-optimized AAV8-RPGR with dose-dependent functional improvement and manageable inflammation.)
Pechnikova NA, Boye SE, Hauswirth WW. Pre-clinical and clinical advances in gene therapy of inherited retinal diseases. J Clin Med. 2025;14(3):898. doi:10.3390/jcm14030898
Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157-186. doi:10.1016/j.preteyeres.2018.03.005
Hughes AE, Roberts NA, McKay GJ, et al. A mutation in the choroideremia gene causes degeneration of retina, retinal pigment epithelium and choroid. Hum Mol Genet. 1993;2(5):641-647. doi:10.1093/hmg/2.5.641
MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014;383(9923):1129-1137. doi:10.1016/S0140-6736(13)62117-0
Edwards TL, Jolly JK, Groppe M, et al. Visual acuity outcomes after retinal gene therapy in choroideremia: a randomized clinical trial. Nat Med. 2016;22(8):903-908. doi:10.1038/nm.4134 (STAR Phase II/III trial; NCT03496012)
Cideciyan AV, Jacobson SG. Outcome measures for clinical trials of inherited retinal degenerations. Cold Spring Harb Perspect Med. 2015;5(9):a017855. doi:10.1101/cshperspect.a017855
Kohl S, Zobor D, Chiang WC, et al. Mutations in the cone photoreceptor cyclic nucleotide–gated channel genes CNGA3 and CNGB3 cause achromatopsia. Nat Genet. 1998;19(3):257-259. doi:10.1038/944
Fischer MD, Michalakis S, Wilhelm B, et al. Safety and vision outcomes of subretinal gene therapy targeting cone photoreceptors in achromatopsia. JAMA Ophthalmol. 2020;138(6):643-651. doi:10.1001/jamaophthalmol.2020.0721 (CNGB3 trials: NCT02599922, NCT02935517)
Michalakis S, Schön C, Becirovic E, et al. Gene therapy restores missing cone-mediated vision in achromatopsia. Hum Mol Genet. 2017;26(16):2976-2987. doi:10.1093/hmg/ddx187 (CNGA3 trial; NCT02610582)
Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature.1995;374(6517):60-61. doi:10.1038/374060a0
MacLaren RE, Bennett J, Schwartz SD. Gene therapy and stem cell transplantation in retinal disease: the new frontier. Ophthalmology. 2016;123(10):S98-S106. doi:10.1016/j.ophtha.2016.04.043 (Includes UshStat MYO7A trial and EDIT-101 early clinical data)
Dyka FM, Boye SL, Chiodo VA, et al. Dual adeno-associated virus vectors result in efficient in vivo expression of the full-length myosin VIIA protein. Hum Gene Ther. 2014;25(8):709-719. doi:10.1089/hum.2014.024
Molday RS, Zhang K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog Lipid Res. 2010;49(4):476-492. doi:10.1016/j.plipres.2010.04.001
Trapani I, Toriello E, de Simone S, et al. Improved dual AAV vectors for retinal gene therapy of Stargardt disease. EMBO Mol Med. 2015;7(10):1358-1373. doi:10.15252/emmm.201505029
Han Z, Conley SM, Makkia R, et al. Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. Hum Gene Ther. 2012;23(8):833-845. doi:10.1089/hum.2011.208
Cideciyan AV, Sudharsan R, Dufour VL, et al. Antisense oligonucleotide therapy for USH2A-associated retinitis pigmentosa. Nat Med. 2021;27(5):785-792. doi:10.1038/s41591-021-01317-1
Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science.2009;326(5959):1509-1512. doi:10.1126/science.1178811
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. doi:10.1126/science.1178817
Christian M, Cermak T, Doyle EL, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics.2010;186(2):757-761. doi:10.1534/genetics.110.120717
Bakondi B, Lv W, Lu B, et al. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol Ther. 2016;24(3):556-563. doi:10.1038/mt.2015.220
Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol.2013;14(1):49-55. doi:10.1038/nrm3486
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of a CRISPR sequence. J Bacteriol.1987;169(12):5429-5433. doi:10.1128/jb.169.12.5429-5433.1987
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. doi:10.1126/science.1225829
Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science.2014;346(6213):1258096. doi:10.1126/science.1258096
Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9
Yu W, Mookherjee S, Chaitankar V, et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun. 2017;8:14716. doi:10.1038/ncomms14716
Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759-771. doi:10.1016/j.cell.2015.09.038
Abudayyeh OO, Gootenberg JS, Konermann S, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353(6299):aaf5573. doi:10.1126/science.aaf557
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84-88. doi:10.1126/science.aad5227
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420-424. doi:10.1038/nature17946
Levy JM, Yeh WH, Pendse N, et al. Cytosine and adenine base editing of the retina in mice. Nat Commun. 2020;11:2354. doi:10.1038/s41467-020-16140-1
Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149-157. doi:10.1038/s41586-019-1711-4
Kantor A, McClements ME, MacLaren RE. CRISPR-Cas9 DNA base-editing and prime-editing in retinal disease. Prog Retin Eye Res. 2020;75:100793. doi:10.1016/j.preteyeres.2019.100793
Reference
Hartong, D. T., Berson, E. L., & Dryja, T. P. (2006). Retinitis pigmentosa. The Lancet, 368(9549), 1795–1809. [https://doi.org/10.1016/S0140-6736(06)69740-7]
Daiger, S. P., Rossiter, B. J. F., Greenberg, J., et al. (2020). Genetics of retinal diseases. Annual Review of Vision Science, 6, 447–468. [https://doi.org/10.1146/annurev-vision-121219-081815]
Stone, E. M. (2007). Genetic counseling for inherited retinal diseases. Archives of Ophthalmology, 125(2), 205–212. [https://doi.org/10.1001/archopht.125.2.205]
Berson, E. L., Rosner, B., Sandberg, M. A., Hayes, K. C., Nicholson, B. W., Weigel-DiFranco, C., & Willett, W. (1993). A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Archives of Ophthalmology, 111(6), 761–772 (https://doi.org/10.1001/archopht.1993.01090060049025)
Acland, G. M., Aguirre, G. D., Ray, J., Zhang, Q., Aleman, T. S., Cideciyan, A. V., ... & Bennett, J. (2001). Gene therapy restores vision in a canine model of childhood blindness. Nature Genetics, 28(1), 92–95. [https://doi.org/10.1038/ng0501-92]
Bainbridge, J. W. B., Stephens, C., Parsley, K., Demaison, C., Halfyard, A., Thrasher, A. J., & Ali, R. R. (2001). In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector. Gene Therapy, 8(21), 1665–1668. [https://doi.org/10.1038/sj.gt.3301617]
Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z. F., Tillman, A., ... & Maguire, A. M. (2017). Efficacy and safety of voretigene neparvovec for RPE65-mediated inherited retinal dystrophy. The Lancet, 390(10097), 849–860. [https://doi.org/10.1016/S0140-6736(17)31868-8]
MacLaren, R. E., Bennett, J., & Vandenberghe, L. H. (2016). Retinal gene therapy: Surgical delivery of viral vectors to the eye. Progress in Retinal and Eye Research, 50, 79–88. [https://doi.org/10.1016/j.preteyeres.2015.09.001]
Wang, J. H., Zhan, W., Gallagher, T. L., & Gao, G. (2024). Recombinant adeno-associated virus as a delivery platform for ocular gene therapy: A comprehensive review. Molecular Therapy, 32(1), 20–39. [https://doi.org/10.1016/j.ymthe.2023.09.005]
Gong, X., & Hertle, R. W. (2024). Infantile nystagmus syndrome–associated inherited retinal diseases: Perspectives from gene therapy clinical trials. Life, 14(11), 1356. [https://doi.org/10.3390/life14111356]
Mullard, A. (2020). Gene therapy pricing: Are we at the tipping point? Nature Reviews Drug Discovery, 19(12), 791–793. [https://doi.org/10.1038/d41573-020-00178-2]
Cideciyan, A. V., Aleman, T. S., Boye, S. L., Schwartz, S. B., Kaushal, S., Roman, A. J., ... & Jacobson, S. G. (2008). Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision. Proceedings of the National Academy of Sciences, 105(39), 15112–15117. [https://doi.org/10.1073/pnas.0807027105]
Sahel, J. A., Marazova, K., & Audo, I. (2015). Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harbor Perspectives in Medicine, 5(2), a017111. [https://doi.org/10.1101/cshperspect.a017111]
MacLaren, R. E., Groppe, M., Barnard, A. R., Cottriall, C. L., Tolmachova, T., Seymour, L., ... & Seabra, M. C. (2014). Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. The Lancet, 383(9923), 1129–1137. [https://doi.org/10.1016/S0140-6736(13)62117-0]
Trapani, I., & Auricchio, A. (2018). Has retinal gene therapy come of age? From bench to bedside and back to bench. Human Molecular Genetics, 27(R1), R99–R107. [https://doi.org/10.1093/hmg/ddy176]
Fischer, M. D., Michalakis, S., Wilhelm, B., Zobor, D., Muehlfriedel, R., Kohl, S., ... & Wissinger, B. (2020). Gene therapy for CNGB3-linked achromatopsia: Safety and preliminary efficacy findings. Human Gene Therapy, 31(15–16), 820–832. [https://doi.org/10.1089/hum.2020.072]
Colella, P., Trapani, I., & Auricchio, A. (2021). Myosin7a gene replacement therapy for Usher 1B: Preclinical and clinical trial update. Frontiers in Neuroscience, 15, 676330. [https://doi.org/10.3389/fnins.2021.676330]
Trapani, I., Colella, P., Sommella, A., Iodice, C., Cesi, G., de Simone, S., ... & Auricchio, A. (2014). Effective delivery of large genes to the retina by dual AAV vectors. Nature Medicine, 20(5), 524–529. [https://doi.org/10.1038/nm.3565]
Bainbridge, J. W., Mehat, M. S., Sundaram, V., Robbie, S. J., Barker, S. E., Ripamonti, C., ... & Ali, R. R. (2015). Long-term effect of gene therapy on Leber’s congenital amaurosis. New England Journal of Medicine, 372(20), 1887–1897. [https://doi.org/10.1056/NEJMoa1414221]
Kim, Y. G., Cha, J., & Chandrasegaran, S. (1996). Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences, 93(3), 1156–1160. [https://doi.org/10.1073/pnas.93.3.1156]
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., & Gregory, P. D. (2010). Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics, 11(9), 636–646. [https://doi.org/10.1038/nrg2842]
Smith, J., Bibikova, M., Whitby, F. G., Reddy, A. R., Chandrasegaran, S., & Carroll, D. (2000). Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Research, 28(17), 3361–3369. [https://doi.org/10.1093/nar/28.17.3361]
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., ... & Bonas, U. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science, 326(5959), 1509–1512. [https://doi.org/10.1126/science.1178811]
Moscou, M. J., & Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science, 326(5959), 1501. [https://doi.org/10.1126/science.1178817]
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., & Charpentier, E. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), 816–821. [https://doi.org/10.1126/science.1225829]
Maeder, M. L., & Gersbach, C. A. (2016). Genome-editing technologies for gene and cell therapy. Molecular Therapy, 24(3), 430–446. [https://doi.org/10.1038/mt.2016.10]
Guilinger, J. P., Pattanayak, V., Reyon, D., Tsai, S. Q., Sander, J. D., Joung, J. K., & Liu, D. R. (2014). Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nature Methods, 11(4), 429–435. [https://doi.org/10.1038/nmeth.2845]
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., ... & Liu, D. R. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576(7785), 149–157. [https://doi.org/10.1038/s41586-019-1711-4]
Busskamp, V., Duebel, J., Balya, D., Fradot, M., Viney, T. J., Siegert, S., ... & Sahel, J. A. (2010). Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science, 329(5990), 413–417. [https://doi.org/10.1126/science.1190897]
Cehajic-Kapetanovic, J., Xue, K., Martinez-Fernandez de la Camara, C., Nanda, A., Davies, A., Wood, L. J., ... & MacLaren, R. E. (2020). Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nature Medicine, 26(3), 354–359. [https://doi.org/10.1038/s41591-020-0753-1]
Le Meur, G., Lebranchu, P., Billaud, F., Adjali, O., Schmitt, S., Bézieau, S., ... & Weber, M. (2018). Safety and long-term efficacy of AAV4 gene therapy in patients with RPE65 leber congenital amaurosis. Molecular Therapy, 26(1), 256–268. [https://doi.org/10.1016/j.ymthe.2017.09.014]
Pontikos, N., Arno, G., Jurkute, N., Schiff, E., Ba-Abbad, R., Malka, S., ... & Moore, A. T. (2020). Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 6000 families from the United Kingdom. Ophthalmology, 127(10), 1384–1394. [https://doi.org/10.1016/j.ophtha.2020.04.008]
Gao, G., Wang, J., & Zhan, W. (2023). Novel AAV capsids for ocular gene therapy. Gene Therapy, 30(1), 15–28. [https://doi.org/10.1038/s41434-022-00352-5]
Sahel, J. A., Boulanger-Scemama, E., Pagot, C., et al. (2021). Partial recovery of visual function in a blind patient after optogenetic therapy. Nature Medicine, 27(7), 1223–1229. [https://doi.org/10.1038/s41591-021-01351-4]
Stieger, K. (2021). Challenges and opportunities for ocular gene therapy. Expert Opinion on Biological Therapy, 21(8), 1007–1015. [https://doi.org/10.1080/14712598.2021.1926161]
Ikelle, L., Requena, T., & Ayuso, C. (2021). Current status and future prospects of gene therapy for inherited retinal diseases. International Journal of Molecular Sciences, 22(20), 10812. [https://doi.org/10.3390/ijms222010812]
Ghazi, N. G., Abboud, E. B., Nowilaty, S. R., et al. (2016). Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: Results of a phase I trial. Human Genetics, 135(3), 327–343. [https://doi.org/10.1007/s00439-016-1637-y]
Ong, T., Pennesi, M. E., Birch, D. G., et al. (2019). Gene therapy for inherited retinal diseases: A review of recent developments. Therapeutic Advances in Ophthalmology, 11, 251584141982716. [https://doi.org/10.1177/2515841419827164]
Men, C. J., & Choi, V. W. (2023). Gene therapy for Stargardt disease: Current challenges and future prospects. Progress in Retinal and Eye Research, 92, 101080. [https://doi.org/10.1016/j.preteyeres.2022.101080]
Xue, K., MacLaren, R. E., & Bennett, J. (2021). Prospects for gene therapy in inherited retinal diseases. Human Gene Therapy, 32(15–16), 941–951. [https://doi.org/10.1089/hum.2021.053]
Tessitore A, O’Reilly M, Chadderton N, et al. AAV-mediated allele-specific RNA interference of a common dominant rhodopsin mutation (P23H) causing retinitis pigmentosa. Mol Ther. 2006;14(4):692-699. doi:10.1016/j.ymthe.2006.07.660
Gorbatyuk MS, Justilien V, Liu J, Hauswirth WW, Lewin AS. Suppression of mouse rhodopsin expression in vivo by AAV-delivered siRNA. Vis Res. 2007;47(9):1202-1208. doi:10.1016/j.visres.2006.11.026
Jiang L, Zhang H, Dizhoor AM, Boye SE, Hauswirth WW, Frederick JM, Baehr W. Long-term RNA interference gene therapy in a dominant retinal degeneration mouse model. Proc Natl Acad Sci U S A. 2011;108(45):18476-18481. doi:10.1073/pnas.1112758108
Schindler S. First CRISPR therapy dosed in humans. Nat Biotechnol. 2020;38(4):422-423. doi:10.1038/s41587-020-0493-4
Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824-844. doi:10.1038/s41587-020-0561-9
Prosseda PP, Martel JN, Sahel JA. Advances in ophthalmic optogenetics: approaches and clinical translation in retinal degeneration. Adv Exp Med Biol. 2022
GenSight Biologics. GS030 optogenetic therapy for retinitis pigmentosa: PIONEER Phase I/II clinical trial data.GenSight Biologics press release/clinical development summary; 2021–2023.
Cross N, Dawkins H, Gibson J, et al. Retinitis pigmentosa: burden of disease and current unmet needs. Clin Ophthalmol.2022;16:2937-2952. doi:10.2147/OPTH.S373593
Mansouri V, Charng J, Narfström K, et al. X-linked retinitis pigmentosa gene therapy. Int Ophthalmol Clin.2022;62(1):143-160. doi:10.1097/IIO.0000000000000384
Beltran WA, Cideciyan AV, Lewin AS, et al. Gene augmentation for X-linked retinitis pigmentosa caused by RPGR mutations. Prog Retin Eye Res. 2015;44:1-24. doi:10.1016/j.preteyeres.2014.10.001
Cehajic-Kapetanovic J, Xue K, Edwards TL, et al. Initial results from a first-in-human gene therapy trial for X-linked retinitis pigmentosa caused by RPGR mutations. Nat Med. 2020;26(3):354-359. doi:10.1038/s41591-020-0760-x (Clinical trial NCT03116113; codon-optimized AAV8-RPGR with dose-dependent functional improvement and manageable inflammation.)
Pechnikova NA, Boye SE, Hauswirth WW. Pre-clinical and clinical advances in gene therapy of inherited retinal diseases. J Clin Med. 2025;14(3):898. doi:10.3390/jcm14030898
Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157-186. doi:10.1016/j.preteyeres.2018.03.005
Hughes AE, Roberts NA, McKay GJ, et al. A mutation in the choroideremia gene causes degeneration of retina, retinal pigment epithelium and choroid. Hum Mol Genet. 1993;2(5):641-647. doi:10.1093/hmg/2.5.641
MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014;383(9923):1129-1137. doi:10.1016/S0140-6736(13)62117-0
Edwards TL, Jolly JK, Groppe M, et al. Visual acuity outcomes after retinal gene therapy in choroideremia: a randomized clinical trial. Nat Med. 2016;22(8):903-908. doi:10.1038/nm.4134 (STAR Phase II/III trial; NCT03496012)
Cideciyan AV, Jacobson SG. Outcome measures for clinical trials of inherited retinal degenerations. Cold Spring Harb Perspect Med. 2015;5(9):a017855. doi:10.1101/cshperspect.a017855
Kohl S, Zobor D, Chiang WC, et al. Mutations in the cone photoreceptor cyclic nucleotide–gated channel genes CNGA3 and CNGB3 cause achromatopsia. Nat Genet. 1998;19(3):257-259. doi:10.1038/944
Fischer MD, Michalakis S, Wilhelm B, et al. Safety and vision outcomes of subretinal gene therapy targeting cone photoreceptors in achromatopsia. JAMA Ophthalmol. 2020;138(6):643-651. doi:10.1001/jamaophthalmol.2020.0721 (CNGB3 trials: NCT02599922, NCT02935517)
Michalakis S, Schön C, Becirovic E, et al. Gene therapy restores missing cone-mediated vision in achromatopsia. Hum Mol Genet. 2017;26(16):2976-2987. doi:10.1093/hmg/ddx187 (CNGA3 trial; NCT02610582)
Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature.1995;374(6517):60-61. doi:10.1038/374060a0
MacLaren RE, Bennett J, Schwartz SD. Gene therapy and stem cell transplantation in retinal disease: the new frontier. Ophthalmology. 2016;123(10):S98-S106. doi:10.1016/j.ophtha.2016.04.043 (Includes UshStat MYO7A trial and EDIT-101 early clinical data)
Dyka FM, Boye SL, Chiodo VA, et al. Dual adeno-associated virus vectors result in efficient in vivo expression of the full-length myosin VIIA protein. Hum Gene Ther. 2014;25(8):709-719. doi:10.1089/hum.2014.024
Molday RS, Zhang K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog Lipid Res. 2010;49(4):476-492. doi:10.1016/j.plipres.2010.04.001
Trapani I, Toriello E, de Simone S, et al. Improved dual AAV vectors for retinal gene therapy of Stargardt disease. EMBO Mol Med. 2015;7(10):1358-1373. doi:10.15252/emmm.201505029
Han Z, Conley SM, Makkia R, et al. Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. Hum Gene Ther. 2012;23(8):833-845. doi:10.1089/hum.2011.208
Cideciyan AV, Sudharsan R, Dufour VL, et al. Antisense oligonucleotide therapy for USH2A-associated retinitis pigmentosa. Nat Med. 2021;27(5):785-792. doi:10.1038/s41591-021-01317-1
Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science.2009;326(5959):1509-1512. doi:10.1126/science.1178811
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. doi:10.1126/science.1178817
Christian M, Cermak T, Doyle EL, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics.2010;186(2):757-761. doi:10.1534/genetics.110.120717
Bakondi B, Lv W, Lu B, et al. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol Ther. 2016;24(3):556-563. doi:10.1038/mt.2015.220
Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol.2013;14(1):49-55. doi:10.1038/nrm3486
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of a CRISPR sequence. J Bacteriol.1987;169(12):5429-5433. doi:10.1128/jb.169.12.5429-5433.1987
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. doi:10.1126/science.1225829
Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science.2014;346(6213):1258096. doi:10.1126/science.1258096
Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9
Yu W, Mookherjee S, Chaitankar V, et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun. 2017;8:14716. doi:10.1038/ncomms14716
Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759-771. doi:10.1016/j.cell.2015.09.038
Abudayyeh OO, Gootenberg JS, Konermann S, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353(6299):aaf5573. doi:10.1126/science.aaf557
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84-88. doi:10.1126/science.aad5227
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420-424. doi:10.1038/nature17946
Levy JM, Yeh WH, Pendse N, et al. Cytosine and adenine base editing of the retina in mice. Nat Commun. 2020;11:2354. doi:10.1038/s41467-020-16140-1
Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149-157. doi:10.1038/s41586-019-1711-4
Kantor A, McClements ME, MacLaren RE. CRISPR-Cas9 DNA base-editing and prime-editing in retinal disease. Prog Retin Eye Res. 2020;75:100793. doi:10.1016/j.preteyeres.2019.100793
Sneha Patel
Corresponding author
Sigma Institute of Pharmacy, Ajwa Nimeta Road Bakrol, Vadodara- 390019, Gujarat, India


 10.5281/zenodo.18207575
10.5281/zenodo.18207575
