Uttaranchal College of Biomedical & Science, Dehradun 248002, Uttarakhand, India
Parkinson's disease (PD) is characterized by progressive neurodegeneration but is also defined by a dynamic and sustained neuroplastic response within the central nervous system. This review explores the dualistic role of neuroplasticity in PD, operating along a continuum from physiological adaptation to pathological dysfunction. Initially, adaptive (compensatory) neuroplasticity—mediated by presynaptic dopaminergic upregulation, postsynaptic receptor super sensitivity, and long-term potentiation (LTP)—preserves motor function, underpinning the prolonged clinical "honeymoon period." However, as neurodegeneration advances and with chronic, pulsatile dopaminergic therapy, these same plastic mechanisms veer into maladaptive (aberrant) territory. Characterized by impaired LTP, enhanced long-term depression (LTD), and pathological circuit reorganization, this shift leads to levodopa-induced dyskinesias, freezing of gait, and other refractory symptoms. Modern neurorehabilitation must therefore strategically guide this inherent plastic potential. This requires adherence to evidence-based principles—specificity, salience, intensity, repetition, and progressive challenge—operationalized through modalities such as aerobic exercise, skill-based training (e.g., LSVT BIG, dance), dual-task practice, and adjunctive technologies (e.g., non-invasive brain stimulation). Effective intervention necessitates a personalized, plasticity-centric paradigm that matches rehabilitation strategies to disease stage, clinical phenotype, and medication state, supported by a multidisciplinary team. Future directions include the development of biomarkers to monitor neuroplasticity, the application of AI for personalization, and novel therapies targeting maladaptive plasticity. Ultimately, a deep understanding of neuroplasticity transforms PD management, positioning targeted rehabilitation not merely as symptomatic care but as a fundamental strategy to promote adaptive rewiring, mitigate pathological dysfunction, and potentially modify disease course.
Neuroplasticity, the central nervous system's dynamic capacity to reorganize its structure, function, and connections in response to experience, injury, or disease, operates along a continuum from physiological to pathological. Physiological neuroplasticity is the foundation of all learning and memory, encompassing synaptic strengthening (long-term potentiation, or LTP) and pruning (long-term depression, or LTD), which allow for the adaptive encoding of new skills and knowledge within healthy neural networks[1]. In contrast, pathological neuroplasticity represents a maladaptive deviation from this normative process, where the brain's rewiring in response to damage or dysfunction generates new, often debilitating, symptoms or perpetuates disease states; this is not merely a failure of plasticity but an active process of erroneous learning and circuit reorganization[2]. This distinction is paramount in Parkinson's disease (PD), where the progressive degeneration of dopaminergic neurons in the substantia nigra triggers a complex and sustained neuroplastic response throughout the brain's motor and non-motor circuits[3][4]. The ensuing cascade of compensatory changes creates what is termed the neuroplastic paradox in PD: the very same adaptive mechanisms that initially preserve function and delay clinical symptom onset can, over time and under the influence of progressive neurodegeneration and chronic dopaminergic therapy, transform into sources of pathological dysfunction and treatment complications[5][6]. Initially, the brain engages in widespread adaptive compensation, such as upregulating dopamine synthesis in remaining neurons, expanding cortical movement representations, and recruiting alternative parallel circuits (e.g., increased reliance on cerebellar and premotor pathways) to maintain motor output despite striatal dopamine depletion, which clinically manifests as the prolonged "honeymoon period" where patients respond well to medication[7][8]. However, as the disease advances, these compensatory efforts become overwhelmed, and the plastic changes veer into maladaptive territory. The chronic, pulsatile replacement of dopamine with medications like levodopa can induce aberrant synaptic plasticity in the striatum, leading to the over-strengthening of direct pathway connections and the generation of involuntary movements known as levodopa-induced dyskinesias. Similarly, the pathological reorganization of sensorimotor circuits contributes to refractory symptoms such as freezing of gait and dystonia, where the brain's internal model for movement becomes fundamentally miscalibrated [9][10]. This paradox underscores that neuroplasticity itself is a neutral tool; its clinical value is determined by the context, timing, and nature of the rewiring it produces[11]. Therefore, the overarching goal of modern neurorehabilitation in PD is to strategically guide this inherent plastic potential toward adaptive outcomes while mitigating maladaptive ones. This requires adherence to several evidence-based principles for harnessing neuroplasticity[12]. The first principle is specificity, meaning that the training provided must directly target the desired functional outcome, as plasticity is experience-dependent; for example, balance training improves balance more than general aerobic exercise alone. The second is salience and attention; the task must be meaningful and engage the patient's focused attention to drive sufficient neuromodulatory (e.g., dopamine, norepinephrine) release to reinforce the correct synaptic connections[13]. The third is intensity and repetition, as robust structural and functional change requires adequate dosing—often quantified as a high number of practice repetitions—to induce and consolidate learning[14][15]. The fourth is progressive challenge, where task difficulty is systematically increased to avoid plateaus and continuously drive adaptation, a concept central to skill-based training like dance or tai chi[16]. Finally, the principle of timing in relation to medication is critical; while training during "ON" periods may allow for greater intensity and motor learning, some evidence suggests that training in the more challenging "OFF" state may preferentially engage and strengthen compensatory pathways. Contemporary rehabilitation modalities are explicitly designed around these principles. Aerobic exercise (e.g., forced-rate cycling) promotes global brain health, increases brain-derived neurotrophic factor (BDNF), and enhances the responsiveness of neural networks to subsequent skill training. High-amplitude, goal-based training (e.g., Lee Silverman Voice Treatment BIG) uses intense repetition of large movements to drive cortical remapping of diminished motor programs[17]. Dual-task and cognitive-motor integration training targets the impaired prefrontal-basal ganglia circuits responsible for automatic movement, thereby addressing freezing of gait. Furthermore, adjuvant technologies like non-invasive brain stimulation (transcranial magnetic stimulation or transcranial direct current stimulation) are being investigated to prime the brain—by modulating cortical excitability and synaptic plasticity—to be more receptive to subsequent behavioral therapy, thereby enhancing the efficacy of rehabilitation[18]. In essence, effective rehabilitation strategy selection in PD moves beyond generic exercise prescription to a targeted, plasticiy-centric paradigm that diagnoses the individual's specific motor and non-motor deficits and then deploys the appropriate combination of salient, intensive, and specific interventions at the optimal time to guide the brain's innate capacity for change toward functional recovery and away from pathological dysfunction.
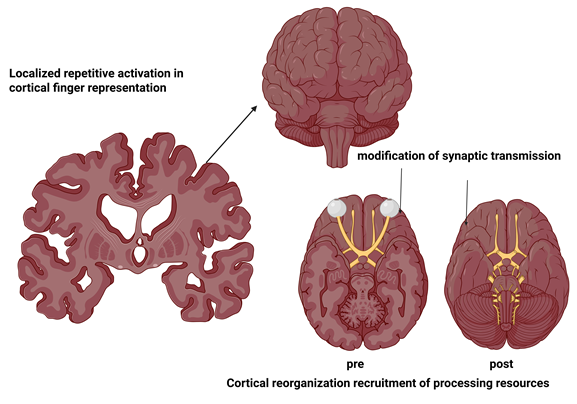
Fig: 1 Neural Mechanisms of Cortical Finger Representation
Adaptive (Compensatory) Neuroplasticity in PD
Cellular and Molecular Mechanisms of Compensation in Parkinson's Disease
The remarkable ability of the Parkinsonian brain to maintain function for years despite the progressive loss of dopaminergic neurons is not a passive process but an active, multifaceted campaign of cellular and molecular compensation[19]. This adaptive neuroplasticity represents the brain's intrinsic attempt to preserve homeostasis within the cortico-striatal-thalamo-cortical motor loops. The compensation occurs at two primary, interconnected levels: presynaptic adaptations within the dwindling population of nigrostriatal neurons, and postsynaptic modifications within the striatal medium spiny neurons (MSNs) and their cortical inputs, primarily centered on the regulation of synaptic strength through mechanisms like long-term potentiation (LTP)[20]. These mechanisms collectively form a biological substrate for the clinical "honeymoon period," delaying the overt manifestation of motor symptoms until a critical threshold of neuronal loss and compensatory failure is reached.
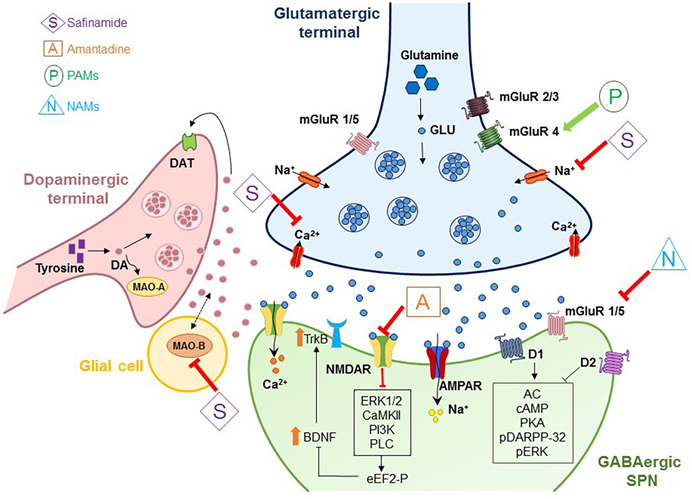
Fig: 2 Striatal Synaptic Plasticity in PD: Molecular Players and Dysregulation
Presynaptic Dopaminergic Adaptation: Maximizing a Diminishing Resource
The initial and most direct line of defense against dopamine depletion is the upregulation of the biosynthetic and functional capacity of the remaining nigrostriatal neurons[21][22]. This is a quintessential example of homeostatic plasticity, where neurons adjust their output to meet network demands despite structural loss. Increased Dopamine Synthesis and Turnover The surviving dopaminergic terminals in the striatum undergo significant biochemical changes. There is a marked upregulation of tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine synthesis[23][24]. This increase in TH activity, and often in the expression of the aromatic L-amino acid decarboxylase (AADC), allows each terminal to produce more dopamine per unit time. Concurrently, the rate of dopamine release and its subsequent reuptake by the dopamine transporter (DAT) accelerates, a state known as increased dopamine turnover. This means the existing dopamine is used and recycled more efficiently to maintain synaptic concentrations[25][26]. This compensatory upregulation is so effective that striatal dopamine levels can remain near-normal until approximately 50-70% of nigral neurons and 70-80% of striatal terminal density are lost, explaining the protracted pre-symptomatic phase of PD. Downregulation of the Dopamine Transporter (DAT) In a seemingly paradoxical but strategic move, the expression and function of DAT on the presynaptic terminal are often reduced[27]. While this might seem counterintuitive for a system trying to conserve dopamine, it serves a critical purpose by slowing reuptake, dopamine molecules spend more time in the synaptic cleft, increasing their probability of binding to postsynaptic receptors[28]. This extends the dopamine half-life and amplifies the signal from each release event, effectively "stretching" the available neurotransmitter. Sprouting and Terminal Arborization There is compelling evidence from animal models and post-mortem human studies suggesting a structural plastic response. Surviving dopaminergic axons may undergo terminal sprouting, increasing the density of their innervation in the striatum to cover a larger territory and form new synaptic connections[30]. This attempt to physically reclaim lost synaptic space represents a more advanced, albeit limited, morphological adaptation. Shift in Firing Patterns The activity of substantia nigra pars compacta neurons also adapts. In addition to the well-known pacemaker firing, there is an increase in phasic or burst firing patterns in response to salient events. Burst firing is associated with a greater release of dopamine per action potential compared to tonic firing, providing a more powerful and behaviorally relevant signal to the striatum to help initiate and guide movement. These presynaptic mechanisms act as a volume knob for the dopaminergic system, turning up the gain on the remaining circuitry. However, this state of hyperfunction is metabolically costly and ultimately unsustainable. The increased oxidative stress associated with elevated dopamine synthesis and turnover may, tragically, accelerate the degeneration of the very neurons fighting to survive, contributing to the eventual collapse of this compensatory phase[31].
Synaptic Strength and Long-Term Potentiation (LTP): Rewiring the Post-Synaptic Response
While presynaptic adaptations focus on neurotransmitter supply, the striatum simultaneously undergoes profound postsynaptic changes to heighten its sensitivity to the available dopamine and reorganize its internal circuitry[32][33]. This is where the classic mechanisms of synaptic plasticity, particularly LTP, come to the forefront. Dopamine Receptor Supersensitivity A cornerstone of postsynaptic compensation is the upregulation and increased sensitivity of postsynaptic dopamine D2 receptors on the MSNs of the indirect pathway. In the healthy state, dopamine binding to D2 receptors inhibits the indirect pathway, which suppresses unwanted movement. With dopamine depletion, this inhibition is lost, leading to excessive indirect pathway activity and movement inhibition (akinesia/bradykinesia). To counteract this, MSNs increase the number and/or coupling efficiency of D2 receptors, making them hyper-responsive to every molecule of dopamine[34][35]. This denervation supersensitivity helps restore the balance between the direct (go) and indirect (stop) pathways. The role of D1 receptors in the direct pathway is more complex, with changes potentially being region-specific and stage-dependent. Altered Threshold for Synaptic Plasticity (Metaplasticity) The fundamental rules governing how synapses strengthen or weaken—their "plasticity threshold"—are changed in the dopamine-depleted striatum[36][37]. This phenomenon, known as metaplasticity, is a higher-order form of plasticity that sets the stage for subsequent learning. Experimental models show that in early PD, the conditions for inducing LTP at corticostriatal synapses are altered. While full dopamine loss ultimately impairs LTP, the partial depletion state during early compensation may shift the balance, potentially lowering the threshold for strengthening synapses that are actively engaged. This metaplastic state may prime the striatum to more readily reinforce alternative motor engrams. The Critical Role of BDNF and Glutamatergic Input Brain-derived neurotrophic factor (BDNF) is a key molecular mediator of activity-dependent plasticity. It is released from cortical neurons projecting to the striatum in response to intense neuronal activity, such as that during motor learning or exercise[38][39]. BDNF binds to TrkB receptors on striatal MSNs, activating intracellular cascades that promote synaptic strengthening, dendritic spine growth, and LTP. In adaptive neuroplasticity, activities that increase cortical BDNF production (like targeted rehabilitation) can help sustain the health and plasticity of striatal neurons, fostering compensatory circuit reorganization. Furthermore, the glutamatergic inputs from the cortex and thalamus are crucial. Compensatory LTP often involves strengthening these excitatory synapses onto MSNs, effectively amplifying the "drive" from cortical motor planning areas to bypass the faltering nigrostriatal gate[40][41].
Clinical Synthesis and Therapeutic Implications
The interplay of these presynaptic and postsynaptic mechanisms creates a fragile but functional equilibrium. Clinically, this is observed as the patient's excellent response to low doses of levodopa in early disease—the supersensitive system reacts powerfully to minimal exogenous dopamine[42][43]. The principles of rehabilitation are designed to engage and support these very molecular processes. High-intensity, skill-based exercise is not merely about fitness; it is a deliberate intervention to drive cortical BDNF release, impose patterned glutamatergic activity on the striatum, and induce LTP-like strengthening in corticostriatal synapses that promote effective movement. Forced-rate exercise, where movement cadence is maintained above a patient's voluntary preference, may be particularly potent in triggering these plasticity-related molecular cascades. Furthermore, engaging in complex, cognitively challenging motor tasks (like dance or tai chi) recruits prefrontal and cerebellar circuits, encouraging the formation of alternative parallel pathways that depend less on the compromised nigrostriatal system[44][45].
Maladaptive (Aberrant) Neuroplasticity in PD
Mechanisms Leading to Dysfunctional Rewiring
The transition from adaptive compensation to maladaptive pathology in Parkinson's disease (PD) is driven by specific mechanisms that corrupt the brain's inherent plasticity. Dysfunctional rewiring occurs when the molecular rules governing synaptic change are distorted and when entire neural circuits lose their equilibrium, leading to the consolidation of pathological patterns rather than functional recovery[46].
Impaired LTP and Enhanced Long-Term Depression (LTD)
At the synaptic level, maladaptive plasticity is characterized by a fundamental corruption of the mechanisms for learning and memory. Dopamine is not merely a modulator of transmission; it is a critical permissive signal for synaptic plasticity. Its severe depletion in the striatum disrupts the delicate biochemical machinery required for long-term potentiation (LTP)—the process that strengthens connections necessary for normal motor learning[47].
Concurrently, the conditions for long-term depression (LTD)—a process that weakens synaptic connections—become pathologically enhanced. This shift occurs because dopamine loss alters intracellular signaling cascades in medium spiny neurons, particularly favoring pathways involving protein phosphatases and endocannabinoid signaling that promote synaptic weakening. The result is a double insult: the brain loses the capacity to strengthen correct, adaptive motor engrams via LTP, while it gains a propensity to permanently weaken those same connections via aberrant LTD. This creates a synaptic environment where pathological circuits, such as those underlying involuntary movements, can be more easily stamped in, as the normal mechanisms for refining and overwriting them are disabled [48].
Disruption of Basal Ganglia Circuitry Balance
The synaptic dysfunction directly manifests as a large-scale disruption in the balance of the basal ganglia's classic direct and indirect pathways. Normally, these pathways work in a dynamic equilibrium to facilitate desired movement and suppress unwanted ones. Dopamine depletion dramatically tilts this balance toward overactivity of the indirect "stop" pathway and underactivity of the direct "go" pathway, leading to akinesia and bradykinesia. However, maladaptive plasticity complicates this picture[49]. Chronic, pulsatile dopamine replacement therapy (e.g., levodopa) can induce aberrant, excessive strengthening of synapses in the direct pathway. This creates a state of pathological hyper-sensitivity, where medication doses cause an overwhelming, dysregulated "go" signal. This is the primary circuit-level mechanism underlying levodopa-induced dyskinesias. Furthermore, progressive neurodegeneration and dysfunctional plasticity in downstream nodes, like the brainstem and pedunculopontine nucleus, contribute to symptoms resistant to dopaminergic therapy, such as freezing of gait. Here, the loss of automatic movement control reflects a failure of integrated circuit processing, not just a simple dopamine deficit [50].
Table 1: Contrasting Adaptive vs. Maladaptive Neuroplasticity in PD
|
Feature |
Adaptive (Compensatory) Neuroplasticity |
Maladaptive (Aberrant) Neuroplasticity |
|
Core Definition & Role |
The brain's beneficial reorganization to preserve function and delay symptom onset despite neurodegeneration. A form of successful compensation. |
The brain's harmful reorganization that generates new dysfunction or perpetuates disease symptoms. A form of pathological learning. |
|
Primary Synaptic Mechanism |
Strengthening of appropriate connections via mechanisms like Long-Term Potentiation (LTP) in compensatory circuits. |
Weakening of useful connections or strengthening of faulty ones. Characterized by impaired LTP and enhanced Long-Term Depression (LTD) in key pathways. |
|
Cellular & Molecular Triggers |
Driven by activity-dependent learning (e.g., exercise, skill practice), BDNF release, and homeostatic responses to partial dopamine loss. |
Triggered by severe, chronic dopamine depletion, pulsatile dopaminergic therapy (e.g., levodopa), and progressive circuit imbalance. |
|
Impact on Basal Ganglia Circuits |
Promotes functional rebalancing: Recruits alternative parallel pathways (cortico-cerebellar, premotor) and increases efficiency in remaining nigrostriatal neurons. |
Causes pathological imbalance: Leads to hyper-sensitivity of the direct pathway and entrenched overactivity of the indirect pathway, disrupting the "go/stop" equilibrium. |
|
Network-Level Effect |
Cortical remapping and expanded representations; increased reliance on cognitive (goal-directed) control of movement. |
Disruption of normal network dynamics; leads to synchronized, oscillatory bursting in beta-band frequencies that inhibits movement. |
|
Key Clinical Manifestations |
The pre-symptomatic phase and the clinical "honeymoon period" where symptoms are mild and responsive to medication. |
Levodopa-Induced Dyskinesias (LID), dystonia, Freezing of Gait (FoG), and treatment-resistant non-motor symptoms (e.g., apathy). |
|
Relationship to Rehabilitation |
The primary target of rehabilitation. Strategies are designed to actively harness and amplify these processes. |
The primary risk to mitigate. Rehabilitation must be carefully timed and structured to avoid reinforcing maladaptive patterns. |
|
Therapeutic Goal |
Harness, promote, and prolong through targeted, intensive, and salient training. |
Prevent, disrupt, or unlearn through medication optimization, specific retraining, and potentially neuromodulation. |
Neuroplasticity-Informed Rehabilitation Strategies
The foundational principles for driving adaptive neuroplasticity in Parkinson's disease (PD) rehabilitation are derived from decades of neuroscience research and form a mandatory framework for translating exercise into meaningful, lasting brain change. These principles are essential because general, casual activity is insufficient to overcome the brain's pathological inertia; intervention must be deliberately designed to force specific neural adaptation. The core principles are specificity, intensity, and repetition. Specificity dictates that the brain improves exactly what it practices. To improve gait, one must practice walking, with a focus on amplitude, rhythm, and balance[51][52]. To improve reaching, one must practice targeted arm movements. This principle targets the remodeling of distinct cortical and subcortical maps for the impaired function. Intensity refers to training at a challenging level, significantly above everyday effort, which is crucial for releasing neurotrophic factors like brain-derived neurotrophic factor (BDNF) that support neuronal survival and synaptic strengthening. Evidence suggests there may be a minimum threshold of intensity (e.g., heart rate or perceived exertion) required to trigger these plasticity mechanisms. Repetition provides the "dose" of practice needed to cement new neural pathways. Hundreds to thousands of correct movement repetitions are often required to induce structural change and automate skills, moving them from conscious, effortful control to more efficient habitual processing—a key deficit in PD.
Equally critical are the principles of task-salience and progressive challenge. Salience means the activity must be meaningful, attention-grabbing, and rewarding to the individual, as engagement directly influences the release of dopamine and other neuromodulators that "stamp in" the learning[53][54]. A boring, repetitive task is less effective than one embedded in an engaging game, social dance, or goal-oriented activity. Progressive challenge ensures continuous adaptation by systematically increasing the task's difficulty, complexity, or cognitive load as the patient improves. This prevents plateaus and forces the nervous system to continually problem-solve and refine motor strategies, thereby expanding the brain's functional capacity and promoting resilience. For instance, balance training progresses from a firm to a soft surface, and then to performing a dual-task while maintaining balance. Together, these principles guide the design of rehabilitation that is not merely symptomatic exercise but a direct, evidence-based intervention to promote adaptive rewiring, combat maladaptive plasticity, and maximize functional independence.
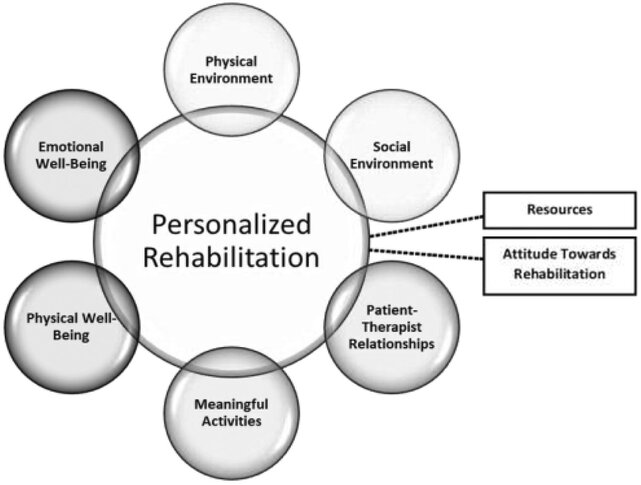
Fig: 3 Exploratory frameworks of personalized rehabilitation.
Modalities to Promote Adaptive Neuroplasticity and Advanced Interventions
The foundational principles for driving adaptive change are operationalized through specific, evidence-based rehabilitation modalities. These interventions are designed to directly target the compromised neural circuits in Parkinson's disease (PD), leveraging the brain's plastic potential to improve function, slow decline, and mitigate maladaptive consequences[56][57].
Modalities to Promote Adaptive Neuroplasticity
Physical Activity and Targeted Exercise
Physical exercise is the cornerstone of neuroplasticity-based rehabilitation, with different forms targeting distinct molecular and systemic pathways.
Aerobic Exercise for BDNF and Network Health Activities like brisk walking, cycling (especially forced-rate), and swimming are potent stimulators of brain-derived neurotrophic factor (BDNF). BDNF is a crucial protein for neuronal survival, synaptic growth, and LTP. Aerobic exercise enhances cerebral blood flow, mitochondrial biogenesis, and overall brain network efficiency[58][59]. Clinically, this translates to improved executive function, processing speed, and mood, which are essential for motor planning and cognitive control of movement. It creates a "primed" brain state that is more receptive to skill-based learning. Resistance Training for Myokines and Strength: While often emphasized for sarcopenia, resistance training has direct neuroplastic benefits. Contracting muscles release myokines, signaling molecules that cross the blood-brain barrier and exert neuroprotective and anti-inflammatory effects. Furthermore, by improving foundational strength, proprioception, and joint stability, resistance training enhances the "signal-to-noise ratio" of motor commands. A stronger, more stable body allows for more precise execution of movements, which provides clearer feedback to the sensorimotor system, facilitating more accurate cortical remapping. Skill-Based and Dual-Task Training (e.g., Tai Chi, Dance): This category most directly applies the principles of specificity, salience, and progressive challenge. Modalities like Tai Chi, tango, or boxing are not merely exercise; they are complex motor-cognitive activities requiring balance, coordination, rhythm, and sequence learning[60][61]. They forcefully engage the prefrontal-striatal-cerebellar networks that are crucial for automatic movement. Dual-task training—such as walking while performing a cognitive task—specifically targets the impaired ability to automate gait, a core deficit in PD that leads to freezing. By practicing under controlled, progressive cognitive load, patients can improve the capacity of their brain to allocate resources and reintegrate divided attention, thereby reducing fall risk and improving gait stability. Cognitive Rehabilitation and Motor-Cognitive Integration Cognitive impairment in PD, particularly in executive function, is a direct result of fronto-striatal circuit dysfunction. Cognitive rehabilitation moves beyond general "brain games" to target specific deficits like set-shifting, working memory, and response inhibition. Techniques include computerized training, strategy-based learning, and, most powerfully, motor-cognitive integration. This involves embedding cognitive challenges directly into physical tasks (e.g., "step onto the tile of a specific color" or "change movement direction on a verbal cue")[62]. This integrated approach forces the weakened prefrontal-basal ganglia connections to be actively engaged and strengthened during real-world, whole-body actions, promoting plasticity in the very circuits needed for functional independence.
Table 2 Neuroplasticity-Focused Rehabilitation Approaches for Parkinson's Disease
|
Rehabilitation Approach |
Primary Neuroplastic Target & Goal |
Key Proposed Mechanisms |
Clinical Evidence & Outcomes |
|
AEROBIC EXERCISE |
Global Network Health & Resilience. |
Increases Brain-Derived Neurotrophic Factor (BDNF), improves cerebral blood flow, reduces neuroinflammation, and promotes mitochondrial health. |
A pivotal study on forced-rate cycling showed a 35% improvement in motor scores (UPDRS-III) after 8 weeks, significantly greater than voluntary exercise. Improves executive function, processing speed, and mood. |
|
RESISTANCE TRAINING |
Structural Maintenance & Synaptic Support. |
Releases muscle-derived myokines (e.g., Irisin) with neuroprotective effects. Improves foundational strength, stabilizing joints to provide clearer proprioceptive feedback to the sensorimotor cortex. |
Linked to improved cognitive control, memory, and gait speed. Prevents sarcopenia, which is crucial for functional independence and fall prevention. |
|
SKILL-BASED & DUAL-TASK TRAINING |
Motor-Cognitive Integration & Cortical Remapping. |
Forcibly engages prefrontal-striatal-cerebellar circuits; requires sequence learning, rhythm, and balance. Enhances sensorimotor integration and challenges divided attention. |
Dance (e.g., tango) significantly improves balance, gait speed, and quality of life. Boxing programs improve bradykinesia, balance, and coordination. Tai Chi reduces fall frequency and improves postural stability. |
|
HIGH-AMPLITUDE TRAINING |
Re-Calibration of Motor Output. |
Uses intensive, high-effort, large-amplitude movements to drive use-dependent cortical reorganization. Teaches the brain to "re-scale" underscaled motor commands. |
Gold-standard evidence: LSVT BIG leads to significant, lasting improvements in movement amplitude, speed, and quality of life. Changes in brain activation patterns on fMRI confirm cortical reorganization. |
|
EXTERNAL CUEING STRATEGIES |
Bypassing Dysfunctional Basal Ganglia. |
Auditory cues (music/metronome) engage cerebellar-auditory-motor networks. Visual cues (lines on floor) shift control to visuomotor parietal pathways. |
Rhythmic Auditory Stimulation (RAS) is a strong evidence-based practice that immediately improves gait speed and stride length, reducing freezing episodes. |
|
COGNITIVE REHABILITATION & MOTOR-COGNITIVE INTEGRATION |
Prefrontal-Striatal Circuits for Executive Function. |
Directly targets and strengthens dorsolateral prefrontal cortex (DLPFC) and its connections to the striatum through structured tasks of attention, memory, and problem-solving. |
Integrated motor-cognitive training (e.g., stepping to cognitive commands) is more effective for improving dual-task walking and reducing fall risk than pure motor or pure cognitive training alone. |
|
NON-INVASIVE BRAIN STIMULATION |
Cortical Excitability & Synaptic Plasticity. |
rTMS (repetitive TMS) induces electrical currents to modulate cortical excitability. tDCS (transcranial DCS) uses weak current to alter neuronal membrane potential, facilitating LTP-like plasticity. |
Used as an adjunct to physical therapy. Anodal tDCS over M1 or DLPFC paired with exercise can enhance motor learning outcomes. rTMS may improve gait, bradykinesia, and depression. |
|
TECHNOLOGY-AUGMENTED THERAPY |
High-Intensity, Salient, Task-Specific Practice. |
VR provides immersive, modifiable environments for safe, graded practice with feedback. Robotics enables massive repetition with perfect consistency and assist-as-needed support. |
VR improves balance, gait, and dual-task performance. Robotic gait training allows for high-dose, high-intensity practice, leading to improvements in walking endurance and quality. |
Personalization and Strategy Selection in Clinical Practice
Assessing Individual Neuroplastic Potential
Currently, there is no single clinical test to quantify a person's "neuroplastic potential." Assessment is indirect, relying on proxies for brain health and responsivity [63][64]. Key factors include disease stage (earlier stages generally retain greater plasticity), cognitive reserve (higher education/ engagement may predict better response to cognitive training), structural brain integrity (seen on MRI as less atrophy or preserved white matter tracts), and genetic profile (e.g., BDNF Val66Met polymorphism can influence plasticity responses). Clinically, a patient's initial rate of motor learning and improvement during early rehabilitation sessions serves as a practical, functional gauge of their current plastic capacity, helping to tailor the intensity and complexity of the program[65][66].
Matching Rehabilitation Strategy to Disease Stage and Phenotype
Personalization requires aligning interventions with the patient's specific challenges. In early-stage PD, the focus is on promoting global brain resilience and building a motor-cognitive reserve through high-intensity aerobic exercise, challenging skill-based training (e.g., dance), and cognitive engagement. For the postural instability/gait difficulty (PIGD) phenotype, the priority shifts to balance training (e.g., Tai Chi), external cueing strategies for freezing, and rigorous dual-task practice. For those with tremor-dominant phenotypes, training may focus more on coordination and dexterity tasks. In advanced stages, the goal becomes maintaining function and independence through compensatory strategy training, safe home exercise routines, and fall prevention[67][68].
Timing Interventions: Medication ON vs. OFF Periods
Strategic timing optimizes outcomes. Training during the medication ON state allows for greater movement amplitude, speed, and safety, facilitating the high-intensity, high-quality practice needed to drive LTP-like plasticity. However, some evidence suggests that practicing in the more challenging "OFF" state may preferentially engage and strengthen compensatory cerebellar and cortical pathways, as the brain is forced to solve motor problems without dopaminergic support. A balanced approach is often best: learning new, complex skills in the "ON" state for optimal performance, while practicing well-learned tasks or cueing strategies in the "OFF" state to improve their robustness and accessibility[69][70].
The Role of Multidisciplinary Care and Patient Engagement
Harnessing neuroplasticity is not solely the domain of physiotherapy. Effective care requires a multidisciplinary team: neurologists to optimize medication timing; physiotherapists for motor training; occupational therapists for functional adaptation; and speech therapists for communication and swallowing. Crucially, long-term adaptive change is impossible without deep patient engagement. Rehabilitation must be personally salient, incorporating patient interests and goals to sustain motivation[71][72]. Educating patients on the "why" behind their exercises—explaining they are actively remodeling their brain—empowers them as active participants in their own care, transforming rehabilitation from a passive prescription into a collaborative, brain-changing endeavor.
Future Directions and Emerging Concepts
Biomarkers of Neuroplasticity for Prognosis and Monitoring
The future of personalized rehabilitation lies in identifying objective biomarkers 9of neuroplasticity. These could include neuroimaging measures (fMRI showing increased connectivity in compensatory networks, MRS for glutamate/ GABA balance), blood-based markers (levels of BDNF, cathepsin B from exercise), and neurophysiological signals (EEG/MEG patterns indicating cortical excitability and plasticity) [73][74]. A key genetic marker is the BDNF Val66Met polymorphism, which affects activity-dependent BDNF release and may predict an individual's responsiveness to exercise-based rehabilitation. Validated biomarkers would allow clinicians to prognosticate recovery potential, objectively monitor the brain's response to therapy, and precisely titrate intervention intensity[75].
The Role of AI and Machine Learning in Personalizing Rehabilitation
Artificial intelligence and machine learning will transform strategy selection by integrating multimodal data—clinical phenotype, biomarker profiles, wearable sensor data on daily movement, and genetic information—to predict optimal rehabilitation pathways. AI algorithms can analyze complex movement patterns from smartphone or sensor data to detect subtle decline or maladaptive patterns (like freezing of gait precursors) in real-time, enabling just-in-time adaptive interventions. Furthermore, AI can dynamically adjust virtual reality or robotic therapy difficulty in response to patient performance, creating a truly personalized and responsive plasticity-enhancing environment [76][77].
Combating Maladaptive Plasticity: Novel Therapeutic Targets
Beyond managing dyskinesias with medication adjustments, future therapies aim to directly correct aberrant synaptic learning[78][79]. Research focuses on pharmacological agents that can normalize the distorted LTP/LTD balance in the striatum without impairing adaptive plasticity, targeting receptors like mGluR5 or NMDA. Advanced neuromodulation, such as closed-loop deep brain stimulation (DBS) that delivers stimulation only when a pathological brain rhythm is detected, may prevent the induction of maladaptive patterns. Targeted sensorimotor retraining protocols, informed by an understanding of distorted body maps, represent a non-invasive behavioral strategy to "unlearn" pathological engrams like dystonia[80].
Promoting Brain Health: The Preventive Potential of Exercise
Perhaps the most powerful application of neuroplasticity principles is primary prevention. In individuals with genetic risk (e.g., LRRK2 carriers) or prodromal signs (REM sleep behavior disorder), structured aerobic and cognitive exercise may bolster brain reserve and cognitive reserve, enhancing network resilience. By upregulating BDNF, reducing neuroinflammation, and improving vascular health, exercise creates a hostile microenvironment for neurodegeneration and a supportive one for adaptive plasticity, potentially delaying symptom onset or slowing progression. This positions prescriptive physical activity not merely as rehabilitation but as a fundamental, disease-modifying strategy in PD management.
CONCLUSION
The journey through Parkinson's disease is fundamentally a story of the brain's struggle to change. Neuroplasticity, the nervous system's inherent capacity for reorganization, emerges not as a mere bystander but as a central protagonist in this narrative. The core paradox of PD is that the very mechanisms which initially sustain function—adaptive neuroplasticity—can, under the strain of progressive degeneration and pharmacotherapy, become the architects of new and debilitating symptoms through maladaptive plasticity. This dichotomy underscores a critical conceptual shift: neuroplasticity itself is neither inherently good nor bad; it is a neutral, powerful tool. The clinical outcome is determined by the context, timing, and direction of the rewiring. This understanding mandates a parallel shift in therapeutic strategy. Rehabilitation in PD must evolve from generic exercise prescription to a targeted, plasticity-informed intervention. The goal is no longer simply to work muscles, but to carefully guide the brain. By rigorously applying the principles of specificity, salience, intensity, and timing, clinicians can design interventions that harness adaptive potential—driving BDNF release, fostering compensatory LTP, and recruiting alternative neural pathways—while avoiding the reinforcement of maladaptive patterns. The integration of advanced tools, from non-invasive brain stimulation to technology-augmented therapy, offers unprecedented precision in delivering this "prescription for plasticity. The path forward is personalized and predictive. Future progress hinges on identifying biomarkers of neuroplasticity to monitor brain change, leveraging AI to tailor dynamic interventions, and developing novel strategies to directly correct aberrant synaptic learning.
REFERENCES

Ruchi Sati, Adaptive and Maladaptive Neuroplasticity in Parkinson's Disease: Implications for Rehabilitation Strategy Selection, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 2904-2923. https://doi.org/10.5281/zenodo.18365964
 10.5281/zenodo.18365964
10.5281/zenodo.18365964
