Adina Institute of pharmaceutical sciences sagar
Background: Microsphere-based drug delivery systems have emerged as a promising technology for enhancing the therapeutic efficacy and safety of pharmaceuticals. These systems utilize biodegradable polymeric microspheres to encapsulate drugs, allowing for controlled and sustained release. This approach improves bioavailability, reduces dosing frequency, and minimizes side effects. Microspheres can be tailored to specific drug properties and target sites, enhancing precision in treatment. This paper reviews recent advancements in microsphere fabrication techniques, including solvent evaporation, spray drying, and coacervation, as well as their applications in treating chronic diseases. Challenges and future directions in optimizing microsphere drug delivery systems are also discussed, highlighting their potential to revolutionize personalized medicine. Methods: Microspheres containing Famotidine drug as a core material were prepared by Solvent Evaporation method. Drugs (Famotidine), HPMC and EC were dissolved in a mixture of ethanol and dichloromethane (1:1) at room temperature (As in table 6). This was poured into 250 mL water containing 0.01% Tween-80 maintained at a temperature of 30–40 °C and subsequently stirred at 300 rpm agitation speed for 45 minutes to allow the volatile solvent to evaporate. The microspheres formed were filtered, washed with water and dried in oven at 37°C (Fartyal et al., 2011). Results: The formulation and characterization of Famotidine loaded microspheres was performed in the present research work. SEM photographs confirmed the shape and formation of the microspheres. The results revealed that experimental conditions allowed a uniform distribution of the biotin compound Famotidine in microspheres having no significant effect on drug-polymer interaction. Finally, the results of this investigation elucidate that the process and formulation variables could be effectively altered to achieve the desired characteristics of the microspheres for novel delivery of the biotin compound. These microspheres are used for drug delivery, wherein the drug can be encapsulated or entrapped. In future it can also be formulated in various dosage forms. Drug: Polymer ratio and Famotidine had a significant effect on % entrapment efficiency, particle size, and % drug release. From the scanning electron microscopy (SEM) study observed that microspheres were spherical and fairly smooth surfaces.
Drug delivery system
Drug delivery systems (DDSs) are pharmaceutical formulations or devices that help in achieving targeted delivery and/or controlled release (CR) of therapeutic agents in our body. Following administration, the DDSs liberate the active ingredients, and subsequently, the bioactive molecules are transported across various biological barriers to reach the site of action. The scientists have contributed substantially to understand the role of different physiological barriers in efficient transport of drugs in the circulatory system and drug movement through cells and tissues. In addition, their significant contribution to the development of a number of new modes of drug delivery has entered clinical practice. Despite a significant advancement in the process of new drug design and discovery, many drugs have unacceptable side effects due to interaction of the drug with parts of the body that are not the target for the drug. Sometimes, side effects occur depending on the medication, the mode of delivery, and response from our body. The buildup of high blood plasma drug concentration due to accumulation of drug from repeated administration of conventional DDS may lead to untoward side effects. Hence, the attempts must be made to afford better patient compliance effect from the reduction in the number and frequency of doses needed to maintain the desired therapeutic responses. These side effects can vary greatly from person to person in type and severity. The method by which the drug is delivered can have a significant impact on its efficacy (Garg et al., 2020)
1.2 Novel drug delivery system (NDDS)
In the past few decades, considerable attention has been concentrated on the evolution of a novel drug delivery system (NDDS) for herbal drugs. Conventional dosage forms, including prolonged-release dosage forms, are unable to satisfy for both holding the drug component at a distinct rate as per directed by the requirements of the body, all through the period of treatment, as well as directing the phytoconstituents to their desired target site to obtain an utmost therapeutic response. In phytoformulation research, developing nano-sized dosage forms (polymeric nanoparticles and nanocapsules, liposomes, solid lipid nanoparticles, phytosomes, and nanoemulsion) has a number of advantages for herbal drugs, including enhancement of solubility and bioavailability, protection from toxicity, enhancement of pharmacological activity, enhancement of stability, improving tissue macrophage distribution, sustained delivery, and protection from physical and chemical degradation. Thus, the nano-sized NDDSs of herbal drugs have a potential future for enhancing the activity and overcoming problems associated with the plant medicines. Liposomes, which are biodegradable and essentially nontoxic vehicles, can encapsulate both hydrophilic and hydrophobic materials (Medina et al., 2004).With the progress in all domains of science and engineering, the dosage forms have developed from simple mixtures and pills to highly sophisticated technology, intensive drug delivery systems, which are known as NDDSs (Bhagwat R et al., 2013).
1.3 Microspheres
Microspheres are polymeric micron range particles with sizes from 1 to 1000 µm. These microspheres are used for drug delivery, wherein the drug can be encapsulated or entrapped form. Based on the polymeric composition of microspheres, they can be classified into two types: natural and synthetic. Natural polymers include carbohydrates (e.g., chitosan, agarose, starch, alginate) and proteins (e.g., albumin, gelatin), while synthetic polymers include nonbiodegradable (e.g., polymethyl methacrylate, epoxy polymers) and biodegradable (polylactic acid/polyglycolic lactic acid) (Prasad, Gupta, Devanna, & Jayasurya, 2014). Microspheres in drug delivery are used for targeted as well as prolonged drug release in the diseased area. It also protects the unstable or pH-sensitive drugs before and after the administration. Microspheres are classified into four different types:

1.4 Types of Microsphere
1. Bioadhesive microspheres
2. Magnetic microspheres
3. Floating microspheres
4. Radioactive microspheres
5. Polymeric microspheres
i) Biodegradable polymeric microspheres
ii) Synthetic polymeric microspheres
1.4.1 Bioadhesive microspheres
These types of microsphere exhibit mucoadhesive property, which allows the drug coated on the surface of the polymer to stick to the targeted organ, resulting in prolonged delivery of the therapeutic agents to the diseased site.
1.4.2 Magnetic microspheres
These microspheres consist of magnetic particles and have a potential to be used for targeted delivery of drugs. Such microspheres can be used for both diagnostic purposes and drug delivery. The drugs encapsulated or coated within these particles can be targeted to the diseased area using an external magnetic source (Joshi et al., 2011). They are utilized very commonly for magnetic hyperthermia in tumor tissues.
1.4.3 Floating microspheres
Floating microspheres are meant to release the drugs loaded in them in gastric content. The bulk density of these drug-loaded microspheres should be kept lower than the gastric juice so that they can float on the surface, thereby having a prolonged drug release.
1.4.4 Radioactive microspheres
Radioactive particles (10–30 µm) are used
for the therapeutic purpose by directly injecting in the veins that are linked to the targeted organ or tissue. These radioactive particles emit three different types of waves: ? emitters, ? emitters, and ? emitters. There are different techniques to prepare such microspheres, and the process of preparation depends on the size of microspheres, route of administration, duration of drug cross-linking, duration of drug release, etc.
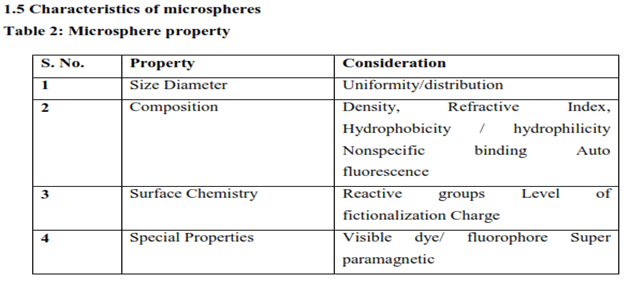
1.6 Application of Microspheres in Pharmaceutical Industry
1.6.1 Microspheres in Vaccine Delivery
The precondition of a vaccine is safety toward the microbes and its harmful components. An ideal vaccine should satisfy this same necessity of effectiveness, protection, affordability in application and charge. The aspect of protection and avoidance of severe effects is complicated. The aspect of safeness and the extent of the manufacturing of antibody responses are intently linked to mode of application. Biodegradable delivery technology for vaccines which are provided by intravenous path may resolve the shortcoming of this same conventional vaccines. The involvement in parenteral (subcutaneous, intramuscular, intradermal) carrier exists even though those who offer significant benefits.
1.6.2 Ophthalmic Drug Delivery
Microspheres developed using polymer exhibits favorable biological behavior such as bioadhesion, permeability-enhancing properties, and interesting physico-chemical characteristics, which make it a unique material for the design of ocular drug delivery vehicles. Eg. Chitosan, Alginate, Gelatin.
1.6.3 Oral drug delivery
The potential of polymer matrix usually contains diazepam like an oral drug delivery has been evaluated through rabbits. Its findings showed that even a film consisting of a 1:0.5 drug-polymer combination may have been an effectual dosage form which is comparable to commercial tablet formulations. The capacity of polymer to establish films could allow use in the formulation of film dosage forms, as an option with drug tablets. The pH sensitivity, combined with both the reactions of the main amine groups, start making polymer a distinctive polymer for oral drug delivery applications.
1.6.4 Gene delivery
Genotype drug delivery involves viral vectors, nonionic liposomes, polycation complexes, and microcapsules technologies. Viral vectors are beneficial for genotype delivery even though those who are extremely efficient and also have a broad variety of cell goals. Even so, if used in vivo they trigger immune responses and pathogenic effects. To resolve the restrictions of viral vectors, nonviral delivery systems have been regarded for gene therapy. Nonviral delivery system does have benefits these as simplicity of preparation, cell / tissue targeting, reduced immune system, unrestricted plasmid size, as well as largescale replicable production. Polymer Will be used as a transporter of DNA for gene delivery applications.
1.6.5 Nasal drug delivery
Polymer based drug delivery systems, such as microspheres, liposomes and gels have been demonstrated to have good bioadhesive characteristics and swell easily when in contact with the nasal mucosa increasing the bioavailability and residence time of the drugs to the nasal route. Eg.Starch, Dextran, Albumin, Chitosan+Gelatin (Virmani and Gupta, 2017).
1.7 Advantages of microspheres
1.8 Disadvantages
Drug discharge from measurements structure differs with an assortment of variables like inalienable and outward factors, food, and the pace of move through the gut The distinction in the delivery rate starting with one portion then onto the next Controlled discharge arrangement normally having a high amount of medications contains veracity and any vanishing of the measurements structure discharge trademark it might show to bring about the development of conceivable poisonousness and treatment disappointment Such measurement structures ought not be compacted or bitten Short drug stacking (limit of half) for the controlled delivery measurement structure, Once managed it is difficult to eliminate the transporter totally from the body, Parental conveyance of microsphere may act together or structure edifices with the blood segment (Kunchu et al;2010)
1.9 Limitation
Some of the disadvantages were found to be as follows (Kunchu et al; 2010).
1. The costs of the materials and processing of the controlled release preparation are substantially higher than those of standard formulations.
2. The fate of polymer matrix and its effect on the environment.
3. The fate of polymer additives such as plasticizers, stabilizers, antioxidants and fillers.
4. Reproducibility is less.
5. Process conditions like change in temperature, pH, solvent addition, and evaporation/agitation may influence the stability of core particles to be encapsulated.
6. The environmental impact of the degradation products of the polymer matrix produced in response to heat, hydrolysis, oxidation, solar radiation or biological agents. (Mohan M et al.,2014)
1.11 Methods used in microsphere preparation
Choosing the method depends primarily on the Character of a polymer been using, the drug, the factors equivocally determined by many formulations and technological factors as the size of the particles requirement, and the drug or protein should not be significantly impacted by the process, the reproducibility of the release profile and the method, there should be no stability Issue, in relation to the finished product. The various types of procedures used to prepare the microspheres using hydrophobic and hydrophilic polymers as matrix materials.
The capacity to integrate medication doses which are relatively small. Stability of preparation after synthesis with a shelf spam which is clinically acceptable.
Controlled particle size and dispensability for injection in the aqueous vehicles. Effective reagent release with strong control over a large time-scale. Biocompatibility of controllable biodegradability and chemical alteration response.
1.11.1 Wax coating and hot melt
Wax used to encapsulate the main components, by dissolving or dispersing the product in melted wax. The waxy paste or mixture, such as frozen liquid paraffin, is released by high intensity blending with cold water. The water is heated up for at least an hour. The substance is stirred up for at least 1 hour. Then the external layer (liquid paraffin) is decanted and the microspheres are immersed in a non-miscible solvent and dry air is required to dry. For the surface ingredients, carnauba wax and beeswax can be used, and both should be combined to obtain desirable characteristics.
1.11.2 Spray drying technique
This was used to prepare polymer microsphere mixed charged with drug. This requires dispersing the raw substance into liquefied coating liquid, and then spraying the mixture into the air for surface solidification accompanied by rapid solvent evaporation. Organic solvent and polymer solution are formulated and sprayed in various weight ratios and drug in specific laboratory conditions producing microspheres filled with medications. This is fast but may lose crystalinity due to rapid drying (Kunchu et al., 2010).

Figure 1
1.11.3 Coacervation
This method is a straight forward separation of macromolecular fluid into two immiscible types of material, a thick coacervate layer, comparatively condensed in macromolecules, and a distilled layer of equilibria. This method is referred to as basic coacervation, in the presence of just one macromolecule. If two or more opposite-charge macromolecules are involved, they are considered complex coacervation. The former is caused by specific factors including temperature shift, Using non-solvent or micro-ions contributing to dehydration in macromolecules, since they facilitate interactions between polymer and polymer through polymer solvent interactions. This can be engineered to generate different properties on microsphere (Kunchu et al; 2010).
1.11.4 Solvent evaporation)
The method of solvent evaporation has also been extensively used to preparation of PLA and PLGA microspheres which contain many various drugs. Several variables were identified that can significantly affect microspheric characteristics, such as solubility of drug, internal morphology, type of solvent, diffusion rate , temperature, polymer composition as well as viscosity, and drug loading. The efficacy of the solvent evaporation system to create microspheres relies on the effective entanglement of the active substance into the particles, and therefore this procedure is particularly efficient with drugs that are either insoluble or partially soluble in the liquid medium that constitutes the constant phase (Kunchu et al., 2010).
1.11.5 Precipitation
It is a modification of the form of evaporation. The emulsion is polar droplets scattered over a non-polar medium. The use of a co-solvent can extract solvent from the droplets. The subsequent rise in the concentration of polymers induces precipitation to create a microspheric suspension (Kunchu et al., 2010)
1.11.6 Freeze Drying
Freeze-drying is effectively used in protein API microspheres preparation. The method is freezing, sublimation, main drying, and secondary drying. At the freezing step, account is taken of the eutectic point of the components. During the process, lyoprotectants or cryoprotectants will stabilize API molecules by removing water, creating a glass matrix, lowering intermolecular interaction by forming hydrogen bonds between the molecules or dipole - dipole interactions. It's a beneficial cycle for heat tolerant molecules, given its high expense. Freeze-drying produces solidification and then enables the reconstitution of particles in an aqueous media (Kunchu et al., 2010).
1.11.7 Single Emulsion Solvent Evaporation Technique
This process requires polymer dissolution in an organic solvent accompanied by emulsification of an aqueous environment containing the emulsifying agent. The resulting emulsion is stirred for several hours in atmospheric conditions to allow the solvent to evaporate, which is then washed, rinsed and dried in desiccators. Designed and manufactured drugs microspheres with polymers by diffusion-evaporation method with emulsion solvent (Kunchu et al., 2010).

Figure 2: single emulsion technique
1.11.8 Double emulsification method
The Doppel-emulsion strategy requires mixing w / o / w or o / w / o processing the double emulsion. The aqueous solution of the product is distributed in a continuous lipophilic organic phase. The continuous step which consists of a polymer solution eventually encapsulates medication Observed in the scattered aqueous layer to form primary emulsion. Prior to introduction to the aqueous solution of alcohol to form primary emulsion, the pre-formed emulsion is subjected to homogenisation or sonication. The microspheres filled with the drug prolonged the release of the medication 24 hours and were Observed to be diffusion and erosion regulated (Kunchu et al .,2010).

Figure 3: Double emulsion technique
1.11.9 Ionic gelation method
Ionotropic gelation is depend on the tendency of polyelectrolytes to cross connect to develop hydrogel beads often called gelispheres in the existence of counter ions. Gelispheres are Circular cross linked polymeric hydrophilic agent capable of substantial gelation and thickening in model biological fluids and drug release regulated by polymer relaxation via it. The hydrogel beads are formed by dumping a drug-laden polymeric solution into the polyvalent cations aqueous solution. The cations migrate through the drug-laden hydrophilic compounds, creating a three-dimensional lattice the moiety is ironically crosslinked. Biomolecules may also be placed into these gelispheres to maintain their three-dimensional form under moderate conditions (Kunchu et al., 2010).
1.12 Material and methods
Table 3
Table 4
1.13 Formulation of microspheres by Solvent Evaporation method
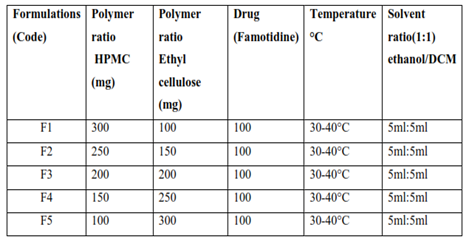
Microspheres containing Famotidine drug as a core material were prepared by Solvent Evaporation method. Drug (Famotidine), HPMC and EC were dissolved in a mixture of ethanol and dichloromethane (1:1) at room temperature (As in table 6). This was poured into 250 mL water containing 0.01% Tween-80 maintained at a temperature of 30–40 °C and subsequently stirred at 300 rpm agitation speed for 45 minutes to allow the volatile solvent to evaporate. The microspheres formed were filtered, washed with water and dried in oven at 37°C (Fartyal et al., 2011).
Table 5: Composition of microsphere formulation
1.4 Evaluation parameter of drug loaded microsphere
1.4.1 Particle size
The particle size is one of the most important parameter for the characterization of microspheres. The size of microspheres was measured using Malvern Zeta sizer (Malvern Instruments). The dispersions were diluted with Millipore filtered water to an appropriate scattering intensity at 25°C and sample was placed in disposable sizing cuvette. The size data is documented in Table 13 (Singh and Vingkar 2008).
1.4.2 Zeta potential
The zeta potential was measured for the determination of the movement velocity of the particles in an electric field and the particle charge. In the present work, the microspheres was diluted 10 times with distilled water and analyzed by Zetasizer Malvern instruments. All samples were sonicated for 5-15 minutes before zeta potential measurements. The zeta potential data is documented in Table 14 (?or?evi? et al., 2015).
1.4.3Quantitative analysis (Entrapment Efficiency)
%Entrapment efficiency was determined by indirect estimation. Drug -loaded microspheres were centrifuged at 15,000 rpm for 30 min using REMI Ultra Centrifuge. The non-entrapped drug (free drug) was determined in the supernatant solution using UV spectrophotometer.
The peak area was determined and amount of free drug is determined by extrapolating the calibration curve. And drug entrapment calculated by using below equation. The entrapment efficiency data is documented in Table 15 (Balla and Goli 2020).
Entrapment efficiency % = Total drug conc. - Supernatant drug conc. / total drug conc.*100
1.4.4 Scanning Electron Microscopic (SEM)
The electron beam from a scanning electron microscope was used to attain the morphological features of the drug loaded microspheres were coated with a thin layer (2–20 nm) of metal(s) such as gold, palladium, or platinum using a sputter coater under vacuum. The pretreated specimen was then bombarded with an electron beam and the interaction resulted in the formation of secondary electrons called auger electrons. From this interaction between the electron beam and the specimen’s atoms, only the electrons scattered at 90° were selected and further processed based on Rutherford and Kramer’s Law for acquiring the images of surface topography (Anwer et al., 2019).
1.5 In-vitro drug release study
The in-vitro drug release study of drug loaded Microsphere formulation was studied by dialysis bag diffusion method. Microspheres were dispersed into dialysis bag and the dialysis bag was then kept in a beaker containing 100 ml of pH 7.4 phosphate buffer. The beaker was placed over a magnetic stirrer and the temperature of the assembly was maintained at 37 ± 2 °C throughout the experiment. During the experiment rpm was maintained at 100 rpm. Samples (2 ml) were withdrawn at a definite time intervals and replaced with equal amounts of fresh pH 7.4 phosphate buffers. After suitable dilutions the samples were analyzed using UV–Visible spectrophotometer at 304 nm. To analyze the in vitro drug release data various kinetic models were used to describe the release kinetics.To analyze the in vitro release data various kinetic models were use to describe the release kinetics. The zero order rate Eq. (2) describes the systems where the drug release rate is independent of its concentration. The first order Eq. (3) describes the release from system where release rate is concentration dependent. Higuchi described the release of drugs from insoluble matrix as a square root of time dependent process based on Fickian diffusion. The results of in vitro release profile obtained for all the formulations were plotted in modes of data treatment.
1.6 Stability studies
The drug loaded Microsphere formulation was packed and were placed in the stability test chamber and subjected to stability studies at accelerated testing (25±2C and 60 ± 5% RH) and (40±2 0C and 70 ±5% RH) for 3 months. The formulation was checked for evaluation parameter particle size, zeta potential and Appearance at the interval of 30, 45, 60, 90 days (3 month) months. The formulation was tested for stability under accelerated storage condition for 3 months in accordance to International Conference on Harmonization (ICH) guidelines.
The formulation was analyzed for the change in evaluation parameter particle size, zeta potential and appearance. All Results were compared against final formulation of 0 days as the reference.
RESULTS AND DISCUSSION
2.1 Evaluation parameter of microsphere formulation
7.7.1 Particle size determination

Figure 4: Particle size (F1)
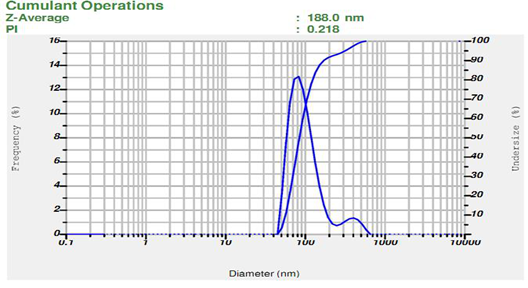
Figure 5: Particle size (F2)

Figure 6: Particle size (F3)

Figure 7: Particle size (F4)

Figure 8: Particle size (F5)
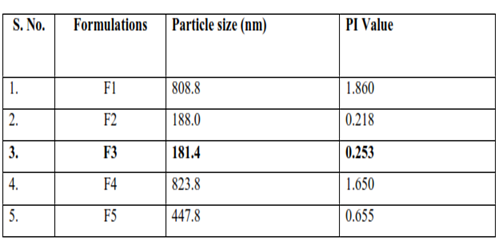
Table 6: Result of Particle size of all formulations
DISCUSSION
The particle size is one of the most important parameter for the characterization of microsphere. The average particle sizes of the prepared microsphere formulation were measured using Malvern zeta sizer. Particle size analysis showed that the average particle size of microsperes was found to be range between 181.4 to 823.8 nm. These particle size values indicate that the all formulated microsphere is under the range of microsphere and F3 is the lowest particle size of all formulation shown in above table
7.7.2 Zeta Potential Determination

Figure 9: Zeta potential (F1)

Figure 10: Zeta potential (F2)

Figure 11: Zeta potential (F3)

Figure 12: Zeta potential (F4)

Figure 13: Zeta potential (F5)
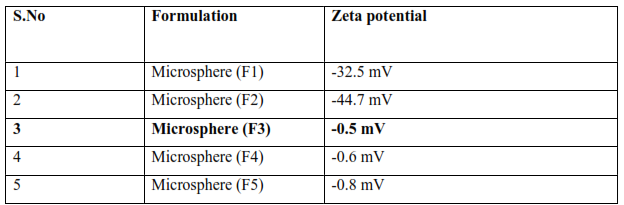
Table 7: Result of Zeta potential of all formulations
DISCUSSION
Zeta potential analysis is carried out to find the surface charge of the particles. The magnitude of zeta potential is predictive of the colloidal stability. Zeta potential was found to be all formulation range -0.5 to -44.7 mV with peak area of 100% intensity. These values indicate that the all formulated microsphere is stable. Results show in above table.
2.3 Entrapment efficacy
Table 8: Entrapment efficacy

DISCUSSION
This might be due to the fact that the variation in entrapment efficiency was due to the changes in the polymer concentration. The prepared drug loaded microsphere formulation possesses high drug entrapment efficiency (90.03) of F3 formulation due to change in polymer concentration and all formulation EE % found to be in the range of 75.99 to 90.03%.
2.4 Scanning electron microscopy characterization of F3 formulation

Figure 14: SEM
DISCUSSION
SEM analysis was performed to determine their microscopic characters (shape & morphology) of prepared microsphere. Drug loaded microsphere were prepared and dried well to remove the moisture content and images were taken using scanning electron microscopy. Scanning electron micrograph of the prepared microsphere at 50.00 kx magnification showed that the microsphere were smooth surface morphology and spherical shape. The smooth surface morphology and spherical shape of microsphere was clearly observed in the SEM images. Show in above figure 18.
2.5 In- vitro drug release kinetics study of F3 formulation
Table 9: Release kinetics study of F3 formulation


Correlation value (R2 value)
Table 10: Correlation value (R2 value)


Figure 15: Zero order model of F3 formulation
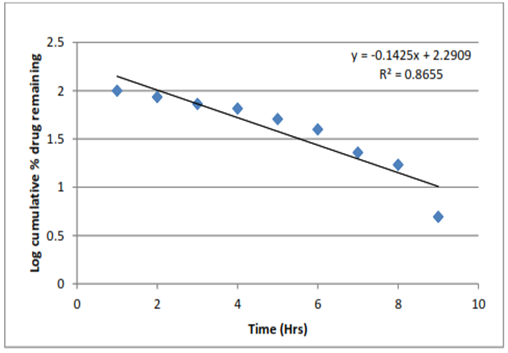
Figure 16: First order model of F3 formulation

Figure 17: Higuchi model of F3 formulation
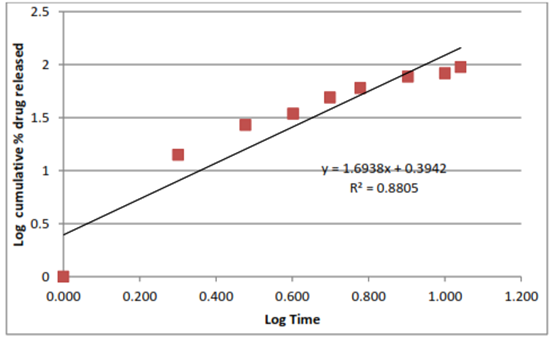
Figure 18: Korsmeyerpeppas
DISCUSSION
The data of percentage drug release formulation were shown in Figure. 19 to 22. For kinetic study following plots were made: cumulative % drug release vs. time (zero order kinetic models); log cumulative % drug remaining vs time (first order kinetic model); cumulative % drug release vs square root of time (Higuchi model); log cumulative % drug release vs log time (Korsmeyer–Peppas model). All Plots are shown in Figure. 19 to 22 and results are summarized in Table 17. Zero order kinetic models refer to the process of constant drug release from a drug delivery device independent of the concentration. The zero order graph of optimized formulation showed the constant drug release from the Formulation, the results of the zero order model was found to be y = 8.776 x + 1.128 R = 0.981. The first order kinetic model describes the release from system where release rate is concentration dependent. The results of first order kinetic model was found to be y = -0.142x+2.290 R?2; = 0.865. The Higuchi model is used to describe the limits for transport and drug release. The Higuchi model of formulation was found to be, y = 30.46x - 15.87 R?2; = 0.906. And the results of Korsmeyerpeppas kinetic model was found to be y = 1.693x + 0.394 R?2; = 0.880. In-vitro drug release was carried out using Franz diffusion cell. The In-vitro drug release data for F3 formulation was performed in pH phosphate buffer 7.4. The F3 formulation showed 95% drug release within 11 hrs. In the above table 14 R2 is correlation value. On the basis of best fit with the highest correlation (R2) value it is concluded that in the F3 formulation of microsphere follow the zero order kinetic model.
2.6. Stability study
Table 11: Stability Study of Microsphere (F3) formulation
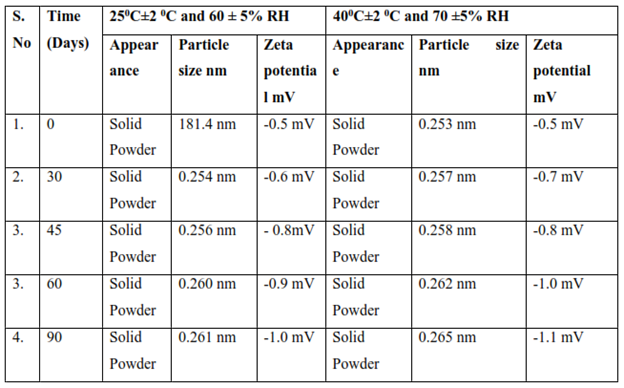
DISCUSSION
Formulation were found to be stable, both physically and chemically, for a period of 3 months at accelerated stability conditions (25C±2C and 60 ± 5% RH) and (40C±2 C and 70 ±5% RH). Physicochemical parameters, including appearance, Zeta potential and particle size were not altered significantly. Results of assay and other evaluation criteria at periodic time points of stability studies are summarized in Table 18.
3.1 SUMMARY AND CONCLUSION
Famotidine has a Characteristic odor and has a solid state and waxy powder form and it was observed that the drug is freely soluble in Glacial acetic acid, soluble in methanol and insoluble in water and ethanol. The melting point of the Famotidine was found to be 163ºC and lambda max of the Famotidine was found to be 304.0 nm. Six points calibration curve were obtained in a concentration range from 10-60 ?g/ml for drug. The response of the drug was found to be linear in the investigation concentration range and the linear regression equation was y = 0.015x + 0.133 with correlation coefficient R?2; = 0.948. Formulation was carried out by Solvent Evaporation method. Trial batches indicated that polymers are suitable for the Famotidine microspheres. Polymer produced good formulations. HPMC and ethyl cellulose were selected for further studies. Scanning electron micrograph of the prepared microspheres at 50.00 kx magnification showed that the microspheres were porous with a smooth surface morphology and spherical shape. The spongy and porous nature of microspheres was clearly observed in the SEM images. Particle size and zeta potential was determined by Malvern Zeta sizer. The particle size analysis confirmed that the prepared sample were in the nanometer range. Average particle size obtained for the formulations F1 and F5 were 181.4 to 823.8 nm. Zeta potential values of microspheres indicated that the formulated microspheres are stable. The amount of drug being entrapped in microspheres was calculated and all the prepared microspheres were found to possess very high entrapment efficiency. The formulation and characterization of Famotidine loaded microspheres was performed in the present research work. SEM photographs confirmed the shape and formation of the microspheres. The results revealed that experimental conditions allowed a uniform distribution of the biotin compound Famotidine in microspheres having no significant effect on drug-polymer interaction. Finally, there results of this investigation elucidate that the process and formulation variables could be effectively altered to achieve the desired characteristics of the microspheres for novel delivery of the biotin compound. These microspheres are used for drug delivery, wherein the drug can be encapsulated or entrapped form. In future it can also be formulated in various dosage forms. Drug: Polymer ratio and Famotidine had significant effect on % entrapment efficiency, particle size, and % drug release. From the scanning electron microscopy (SEM) study observed that microspheres were spherical and fairly smooth surfaces.
REFERENCE

Ambika Chaurasia*, Development And Characterisation Of Floating Microspheres Of Famotidine For The Treatment Of Peptic Ulcer, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 7, 2058-2083. https://doi.org/10.5281/zenodo.13123187
 10.5281/zenodo.13123187
10.5281/zenodo.13123187
