1Department of Pharmaceutical Technology, Raghu College of Pharmacy, Visakhapatnam, A.P., India.
2,3,4,5Department of Pharmaceutical Technology, Sri Venkateswara College of Pharmacy, Srikakulam, A.P., India.
Objective: The present study was aimed to formulate and characterize press coated pulsatile release tablets of Ranitidine hydrochloride in order to obtain a time controlled release for the treatment of nocturnal acid breakthrough. Methods: The tablets were prepared by three different steps which included preparation of floating core tablets with different osmogens (sodium chloride, potassium chloride, dextrose) with different concentrations (1:0.125, 1:0.25, 1:0.5), followed by time-lagged (4 hrs) coating with hydrophobic rupturable polymer ethyl cellulose and finally compression coating with immediate release dose of ranitidine hydrochloride and supporting buoyant layer. Results: Differential scanning calorimeter (DSC) and Fourier transform infrared spectroscopy (FTIR) study showed compatibility between the drug and the polymers used. All the preliminary trials were characterized for different post compression parameters like thickness, hardness, weight variation, friability and drug content along with in vitro dissolution testing.Conclusion: The release profile of optimized tablets showed an immediate release of 99.67% of the drug for 1hr from the outer layer followed by a lag time of 4hrs. The core tablets then released 98.97% of the drug over a period of 12hrs. Hence such a drug delivery system delivered drug at the time when it was needful to the patients suffering from duodenal ulcers..
Pulsatile drug delivery systems deliver drug at a specific time interval as per the requirement of pathophysiological conditions of the disease and thus show better patient compliance. These systems are especially advantageous for the drugs exhibiting chronopharmacological behavior [1]. The availability of various amounts of drug at predictably different times within the circadian cycle results in enhanced therapeutic efficacy and minimizes undesired effects of drug [2]. Diseases like rheumatic arthritis, hypercholesterolemia, bronchial asthma, angina pectoris, myocardial infarction, ulcers and hypertension show symptomatic changes due to circadian rhythms [3]. The advantage of pulsatile release system over the conventional immediate release tablets is their efficiency in properly controlling the drug concentration in the plasma and avoidance of repetitive dosing [4]. An ulcer is a crater like lesion in a membrane and those which develop in areas of the GIT exposed to acidic gastric juices are called peptic ulcers [5]. A peptic ulcer is a sore or lesions in the lining of first part of small intestine, the duodenum. Here, mucous membrane lining of the gastro intestinal tract erodes and causes a steady tissue breakdown. The most common symptom of a peptic ulcer is a burning stomach pain is [6]. Helicobacter pylori are gram-negative bacteria that selectively colonize in the gastric epithelium. It sticks to cells with the aid of specific adhesion molecules and once attached, they deliver damaging toxins to the mucosa [7]. Ranitidine hydrochloride (N, N dimethyl-5-[2-(1- methylamine-2-nitrovinyl)-ethyl thio methyl] furfurylamine hydrochloride) is a selective, competitive H2-receptor antagonist which is widely used in short term treatment of duodenal ulcers and in management of gastric hypersecretory conditions. It is used in combination with proton pump inhibitors for the treatment of night time heart burn [8-10]. The objective of the current study was to evaluate the in vitro performance of press coated pulsatile release tablet formulation of ranitidine hydrochloride as a model drug in the chronotherapy of nocturnal acid breakthrough. The formulated tablets were evaluated for various tablet parametric tests like hardness, thickness, weight variation, friability, drug content, buoyancy and in vitro drug release. The in vitro drug release data was fitted to different kinetic models like zero order, first order, Higuchi, Hixson–Crowell and Korsmeyer–Peppas for studying the mechanism of the drug release from the formulation.

Fig. 1: Structure of ranitidine HCl
Ranitidine hydrochloride, HPMC K4M, crospovidone were obtained from Yarrow chem products, Mumbai. Sodium bicarbonate, sodium chloride were obtained from Finar limited, Gujarat. Ethyl cellulose, HPMC E15, potassium chloride, dibutyl pthalate, iso propyl alcohol were obtained from Fisher scientific, Mumbai.
2.2. Preparation of core tablets
The core tablets of ranitidine hydrochloride were prepared by direct compression method according to the formula give in Table 1. Ranitidine hydrochloride was mixed with HPMC K4M, sodium bicarbonate for 10 min and passed through #30 sieve. Osmogens like NaCl, KCl and dextose, aerosil as a binder were added in geometric dilution and mixing was continued for another 10 min. This mixture was then passed through #60 mesh sieve and magnesium stearate, micocrystalline cellulose were added and mixing was continued for another 10 min. The blend was then compressed into tablets having average weight 300 mg using a single station tablet punching machine fitted with 10 mm round standard concave punches [11].
Table 1: Composition of core formulations of ranitidine hydrochloride
|
Ingredients |
F1 mg |
F2 (1:0.125) mg |
F3 (1:0.25) mg |
F4 (1:0.5) mg
|
F5 (1:0.125) mg |
F6 (1:0.25) mg |
F7 (1:0.5) mg |
F8 (1:0.125) mg |
F9 (1:0.25) mg |
F10 (1:0.5) mg |
|
Ranitidine hydrochloride |
75 |
75 |
75 |
75 |
75 |
75 |
75 |
75 |
75 |
75 |
|
HPMC K4M |
30 |
30
|
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
|
NaHCO3 |
35 |
35 |
35 |
35 |
35 |
35 |
35 |
35 |
35 |
35 |
|
NaCl |
- |
9.3 |
18.7 |
37.5 |
- |
- |
- |
- |
- |
- |
|
KCl |
- |
- |
- |
- |
9.3 |
18.7 |
37.5 |
- |
- |
- |
|
Dextrose |
- |
- |
- |
- |
- |
- |
- |
9.3 |
18.7 |
37.5 |
|
Aerosil |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
|
Magnesium stearate |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
3.5 |
|
Microcrystaline cellulose |
153 |
143.6 |
134.2 |
115.5 |
143.6 |
134.2 |
115.5 |
143.6 |
134.2 |
115.5 |
|
Total |
300 |
|||||||||
Infrared (IR) spectra of drug, HPMC K4M, ethyl cellulose, crushed tablets were performed for detection of any interactions between the drug and the polymer in the range of 400cm-1 to 4000cm-1 by using ZnSe engine diamond ATR model (Agilent, cary 630) and studied for the presence of characteristic peaks [12].
2.2.2. Differential scanning Calorimetry
The DSC analysis of pure drug, polymers, crushed tablets were carried out to evaluate any possible drug polymer interaction. The analysis was performed at a rate 10 0C/min from 20 0C to 300 0C temperature range under nitrogen flow of 25 mL/min [13].
2.3. Determination of micromeretic properties
The tablet powder mix was characterized for bulk density, tapped density, compressibility index, Hausner’s ratio and angle of repose [14].
2.4. Determination of post compression parameters
2.4.1. Hardness: This test was performed to check the hardness of tablets as they may undergo chipping or breakage during storage, transportation and handling. Six tablets were selected randomly and the hardness of every tablet was measured using a Monsanto hardness tester.
2.4.2. Friability: Roche friabilator was used to perform the friability test where the percentage loss in weight of the tablets or friability (F) was calculated by the formula
F= (1-W/W0) × 100
F=Friability; W0= Initial weight; W=Final weight
2.4.3. Weight variation: To check the uniformity of weight of the tablets, this test was performed by finding the weight of 20 random tablets and calculating their average weight to know the deviation in weight of the tablets.
Initial weight-Final weightInitial weight
2.4.4. Drug content: This test was performed by taking twenty tablets randomly which were weighed and made into a fine powder. A quantity of powdered tablet equal to 75 mg of Ranitidine hydrochloride was dissolved in 0.1 N HCl in 100 ml volumetric flask. It was diluted and absorbance was measured at 314 nm using 0.1 N HCl as a blank [15].
2.4.5. In vitro buoyancy determination:
Here both the floating lag time and total floating time of the tablets was determined.
2.4.6.In vitro drug release studies of core tablets:
In vitro drug release studies of core tablets were carried out for 4 hrs using basket type dissolution apparatus (USP-XXIII Electrolab, Mumbai) containing 900 ml of dissolution apparatus 37±0.5 0C and speed of agitation at 50 rpm. The ranitidine hydrochloride tablets were suspended in dissolution medium consisting 900 ml of gastric (0.1 N HCl) fluid and the process were continued for up to 4 hrs. At predetermined intervals of time, 5 ml aliquots were withdrawn and replaced with an equal quantity of fresh and pre warmed dissolution medium. After suitable dilutions, the samples were analyzed spectrophotometrically at 314 nm. From the drug release data, optimized core tablet formulation was selected and this was further coated to prolong the drug release for a period of 12 h [16].
2.5. Coating of optimized core tablets
2.5.1. Preparation of coating solution
5% w/w coating solutions of ethyl cellulose along with HPMC as erodible polymer and sodium bicarbonate were prepared in isopropyl alcohol and water. The weight ratios of ethyl cellulose to HPMC (Methocel E15) were considered to be 60:30 % and 90:10 %. The solution was plasticized with dibutyl pthalate (20% w/w, with respect to dry polymer), and then talc was added as glidant (5% w/w, related to dry polymer). This formed homogeneous dispersion was stirred gently throughout the process of coating[17]. The polymer solution was sprayed on to the core tablets in conventional pan coating apparatus maintained at 60 0C 50rpm till the desired weight gain (5%, 7.5%, and 10%) was achieved. At each stage the coated tablets were further dried in the coating pan for 15 min at 60 0C and were then placed in the oven at 40 0C for 2 h to remove residual solvent if any[18].
Table 2: Composition of coating solution
|
S. No |
Ingredients |
|
1 |
Ethyl cellulose (5% w/w with respect to dry polymer) |
|
2 |
HPMC E15 (5% w/w) |
|
3 |
Sodium bicarbonate (NaHCO3) (5% w/w) |
|
4 |
Dibutyl phthalate (20% w/w with respect to dry polymer) |
|
5 |
Isopropyl alcohol |
|
6 |
Talc (5% w/w related to dry polymer) |
2.5.2. In vitro drug release studies for coated tablets
In vitro drug release studies were carried out by using paddle type dissolution apparatus (USP-XXIII Electrolab, Mumbai) containing 900 ml of 0.1N HCl maintained at 37±0.5 0C and 50rpm speed upto 12 hrs. At predetermined intervals of time, 5 ml aliquots were withdrawn and replaced with an equal quantity of fresh and pre warmed dissolution medium. After suitable dilutions, the samples were analyzed spectrophotometrically at 314 nm.
2.6.Preparation of immediate release tablets
Immediate release tablets were prepared by direct compression method using different concentrations of super disintegrant crospovidone. All the ingredients in table 3 was accurately weighed and to this sodium bicarbonate was added as a gas generating agent, passed through sieve no. 20 and thoroughly blended.
Table 3: Composition of compression coating with buoyant immediate dose layer
|
S. No |
Ingredients |
I1 |
I2 |
I3 |
|
1 |
Ranitidine hydrochloride |
75 |
75 |
75 |
|
2 |
Crospovidone |
- |
5 |
10 |
|
3 |
NaHCO3 |
40 |
40 |
40 |
|
4 |
Magnesium stearate |
15 |
15 |
15 |
|
5 |
Microcrystalline cellulose |
70 |
65 |
60 |
|
Total |
200 mg |
|||
2.6.1. Preparation of press coated tablets
Press coated tablets were prepared by taking optimized coated core tablet as inner layer and optimized immediate layer tablet as outer layer.
2.7.1. Formulation of floating osmotic tablets of ranitidine hydrochloride with immediate release dose
50% w/w (of the tablet) of the immediate release layer powder was added to the die cavity and then optimized floating osmotic release tablet was placed exactly at the center of the die on the powder. To it remaining 50% w/w (of tablet) of the immediate release layer powder was added in such a way that the osmotic release core tablet was fully covered. It was later compressed with a multi station tablet punching machine using 12 mm punches.
2.7.2. In vitro drug release of ranitidine hydrochloride floating osmotic tablets with immediate release dose
In vitro drug release studies were carried out by using paddle type dissolution apparatus (USP-XXIII Electrolab, Mumbai) containing 900 ml of 0.1N HCl maintained at a temperature of 37±0.5 0C and speed of agitation at 50 rpm. At predetermined intervals of time, 5 ml aliquots were withdrawn and replaced with an equal quantity of fresh and pre warmed dissolution medium. The samples were analyzed spectrophotometrically at 314 nm. After 1hr the tablets were placed in another basket with 900 ml of dissolution fluid maintained at 37±0.5 0C and speed of agitation at 50 rpm for 12 hrs. The above process was repeated.
RESULTS AND DISCUSSION
Floating osmotic tablets of ranitidine hydrochloride using three different osmogens were evaluated using various tests and the results obtained were reported here.
Compatibility studies
FTIR spectra was recorded to assess the compatibility of the drug and excipients. From the IR spectra of drug-polymer mixture, it was found that there occurred no major shift in the frequencies of the characteristic functional groups of the drug. Absence of significant shifting in intensity of peaks and band width showed that no incompatibilities occurred between the drug and polymers used.
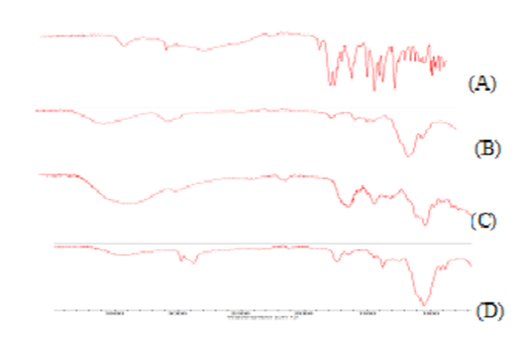
Fig. 2: FTIR overlay spectrogram of (A) Ranitidine hydrochloride, (B) HPMC K4M, (C) CF5, (D) Ethyl cellulose
The DSC thermogram of ranitidine hydrochloride exhibited a sharp endothermic peak at 147.20 0C corresponding to its melting point. The DSC curves of HPMC K4M and ethyl cellulose were obtained at 92.55 0C and 174.76 0C corresponding to their respective melting points. In the DSC curve of optimized formulation, the original peaks were retained and no additional peaks were obtained. This indicated that ranitidine hydrochloride and the polymers used are compatible with each other.
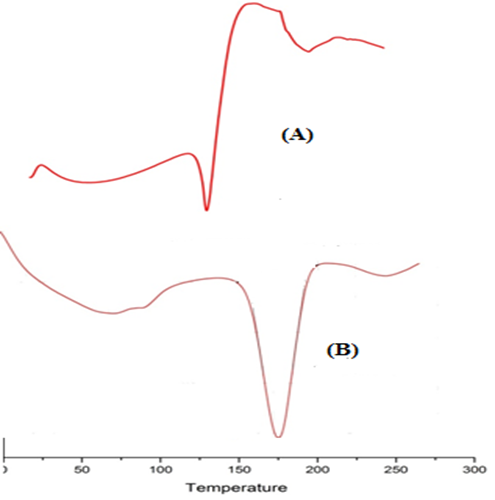
Fig. 3: DSC thermogram overlay of (A) Ranitidine HCl; (B) CF5 formulation
Micromeretic properties
Angle of repose of core formulations was found to be 18.68-23.47o and immediate release formulations was found to be 18.92-19.46 o. Bulk density of core formulations was found to be 0.32-0.4 g/cm3 and immediate release formulations was found to be 0.36-0.47 gm/cm3. Tapped density of core formulations was found to be 0.42-0.57 g/cm3 and immediate release formulations was found to be 0.46-0.52 g/cm3. Hausner's ratio of the core formulations was found to be 0.101-1.60 and immediate release dose formulations was found to be 1.106-1.333. Carr's index of core formulations was found to be 19.23-37.735 and immediate release dose formulations was found to be 9.615-15.217. The results are shown in Table 4.
Table 4: Micromeretic properties of core and immediate release formulations
|
Formulation codes |
Angle of repose (?0) |
Bulk density (g/cm3) |
Tapped density (g/cm3) |
Hausner's ratio |
Carr's index (%) |
|
F1 |
18.99±0.94 |
0.34±0.12 |
0.46±1.18 |
1.36 |
27.35 |
|
F2 |
19.80±0.63 |
0.36±0.25 |
0.48±0.89 |
1.33 |
25 |
|
F3 |
21.38±0.6 |
0.36±1.12 |
0.47±0.03 |
1.30 |
23.4 |
|
F4 |
18.68±1.2 |
0.33±0.69 |
0.44±2.0 |
1.333 |
25 |
|
F5 |
19.75±0.8 |
0.33±0.99 |
0.53±1.29 |
1.60 |
37.735 |
|
F6 |
19.74±0.2 |
0.34±0.32 |
0.5±0.70 |
1.44 |
32 |
|
F7 |
22.96±0.10 |
0.38±0.72 |
0.57±0.59 |
1.502 |
33.450 |
|
F8 |
23.47±0.65 |
0.32±0.06 |
0.42±0.43 |
0.101 |
23.990 |
|
F9 |
20.60±0.37 |
0.4±1.16 |
0.5±0.92 |
1.25 |
20 |
|
F10 |
19.76±0.19 |
0.34±1.65 |
0.42±0.29 |
1.238 |
19.23 |
|
I1 |
18.92±0.32 |
0.39±0.52 |
0.46±0.16 |
1.17 |
15.217 |
|
I2 |
19.46±0.21 |
0.47±0.61 |
0.52±0.64 |
1.106 |
9.615 |
|
I3 |
18.99±0.66 |
0.36±0.49 |
0.48±0.58 |
1.333 |
25 |
Post compression parameters
Hardness of core formulations was found to be in the range of 5.5-7.7 kg/cm2 and immediate release formulations found to be 5.2-6.0 kg/cm2. Weight of the tablets was found to be in the range of ±5% as per limits of Indian Pharmacopoeia. The drug content of the core formulations was found to be 97.89-100.34% and immediate release formulations found to be 88.45-100.23%. Friability of the core formulations was found to be 0.423-0.592% and immediate release dose was found to be 0.231-0.436%. The results are shown in Table 5
Table 5: Post compression parameters of core and immediate release formulations
|
Formulation code |
Thickness (mm) |
Hardness (kg/cm3) |
Weight variation |
Friability (%) |
Drug content (%) |
|
F1 |
3.90±0.12 |
6.8 |
300±0.16 |
0.503 |
99.89±1.75 |
|
F2 |
3.85±0.54 |
6.5 |
299±1.11 |
0.543 |
99.93±0.71 |
|
F3 |
3.89±0.21 |
6.0 |
300±0.02 |
0.488 |
98.10±0.12 |
|
F4 |
3.10±0.64 |
7.7 |
299±0.07 |
0.470 |
97.89±0.19 |
|
F5 |
2.98±0.10 |
5.9 |
299±0.65 |
0.529 |
99.12±0.33 |
|
F6 |
3.65±0.35 |
6.2 |
300±0.14 |
0.423 |
98.45±0.67 |
|
F7 |
3.70±0.19 |
7.4 |
299±0.43 |
0.534 |
100.34±0.58 |
|
F8 |
2.95±0.28 |
6.0 |
298±0.87 |
0.592 |
99.37±0.97 |
|
F9 |
3.56±0.81 |
6.4 |
299±0.17 |
0.567 |
98.56±0.35 |
|
F10 |
3.20±0.21 |
6.3 |
299±0.19 |
0.489 |
99.67±0.21 |
|
I1 |
3.12±0.50 |
5.6 |
200±0.21 |
0.269 |
100.23±0.67 |
|
I2 |
3.14±0.18 |
5.2 |
199±0.19 |
0.231 |
99.90±0.58 |
|
I3 |
3.02±0.32 |
6.0 |
199±0.09 |
0.436 |
98.45±0.34 |
In vitro buoyancy determination:
All the formulations of core tablets floated for a period of 24 hrs when dispersed in 0.1 N HCl solution and immediate release tablets for a period of 4hrs.
Table 6: In vitro buoyancy
|
Formulation code |
Floating lag time (sec) |
Total floating time (hrs) |
|
F1 |
40 |
24 |
|
F2 |
45 |
24 |
|
F3 |
55 |
24 |
|
F4 |
95 |
24 |
|
F5 |
52 |
24 |
|
F6 |
97 |
24 |
|
F7 |
63 |
24 |
|
F8 |
52 |
24 |
|
F9 |
44 |
24 |
|
F10 |
51 |
24 |
|
I1 |
10 |
4 |
|
I2 |
20 |
4 |

Fig. 4: Stages of floating for F2 formulation
In vitro drug release studies of core tablets:
Formulation F1 was prepared without osmogen. The drug release rate was found to be 96.23% respectively in 150 min. Formulation F2 was prepared by adding sodium chloride (1:0.125). The drug release rate was found to be 96.45% respectively in 180 min. Formulation F3 was prepared by adding sodium chloride (1:0.25). The drug release rate was found to be 98.76% respectively in 210 min. Formulation F4 was prepared by adding sodium chloride (1:0.5). The drug release rate was found to be 98.72% respectively in 240 min.
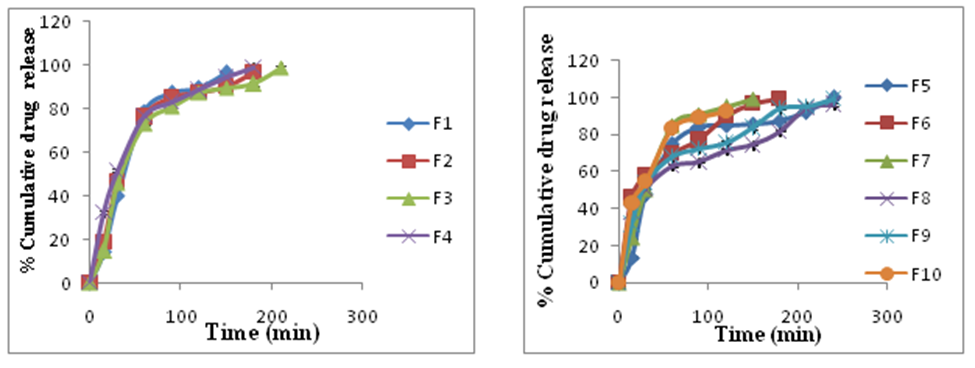
Fig. 5: In vitro dissolution studies of ranitidine hydrochloride floating core tablets F1 to F10
Formulation F5 was prepared by adding potassium chloride (1:0.125). The drug release rate was found to be 99.72% respectively in 180 min. Formulation F6 was prepared by adding potassium chloride (1:0.25). The drug release rate was found to be 99.53% respectively in 180 min. Formulation F7 was prepared by adding potassium chloride (1:0.5). The drug release rate was found to be 99.10% respectively in 150 min. Formulation F8 was prepared by adding dextrose (1:0.125). The drug release rate was found to be 96.43% respectively in 240 min. Formulation F9 was prepared by adding dextrose (1:0.25). The drug release rate was found to be 99.10% respectively in 210 min. Formulation F10 was prepared by adding dextrose (1:0.5). The drug release rate was found to be 92.72% respectively in 120 min.
In vitro drug release studies for coated tablets of ranitidine hydrochloride
From the in vitro drug release studies of coated tablets 10% of CF5 gave about 99.10% of drug release for 12 hrs and CF9 gave about 99.28% of drug release for 12 hrs. From the dissolution studies we can say that increasing in % weight gain decreases the % drug release. So CF5 and CF9 - 10% were optimized for compression coating of coated tablet with an immediate release dose

Fig. 6: Comparative drug release profiles of ranitidine hydrochloride floating osmotic coated tablets CF5 and CF9
In vitro drug release data of outer layer of immediate release tablets
Formulation I1 is prepared without adding super disintegrant. The drug release rate was found to be 78.16% respectively in 1 hr. Formulation I2 and I3 were prepared by adding super disintegrant. The drug release rate was found to be 82.8% and 99.67% respectively in 1 hr.

Fig. 7: Comparative drug release profiles of immediate release dose
Compression coating of coated tablets of ranitidine hydrochloride with immediate release (I3)
Post compression parameters of press coated tablets
Hardness of the formulations was in the range of 6.2-6.6 kg/cm2. Weight variation of the tablets was found to be in the range as per the limits specified in pharmacopoeia. Percent friability of the prepared tablets was found to be less than 1?viation and the drug content was in the range of 99.56-100.01.
Table 7: Post compression parameters of press coated tablets
|
Formulation code |
Thickness (mm) |
Hardness (kg/cm3) |
Weight variation |
Friability (%) |
Drug content (%) |
|
CF5 |
4.10±0.02 |
6.2 |
550±0.05 |
0.61 |
99.56±1.42 |
|
CF9 |
4.12±0.04 |
6.6 |
549±0.08 |
0.59 |
100.01±1.23 |
In vitro drug release study
From in vitro drug release studies for compression of coated tablets of ranitidine hydrochloride with an immediate release (I3), CF5 gave 98.97% drug release for 12 hrs and CF9 gave 86.55% drug release for 12 hrs.
Drug release kinetics
Kinetics were applied to optimized floating core tablets because there is no drug release for certain time in floating osmotic tablets. Correlation coefficients of all the prepared formulations showed greater values with zero order plots than first order. To confirm the drug release mechanism from these tablets, the data were fitted to Higuchi and Korsemeyer-Peppas equation. The two optimized formulations showed higher regression values for Korsemeyer-Peppas. According to Peppas kinetics based on n value the mechanism of drug release was non fickian or anomalous transport.
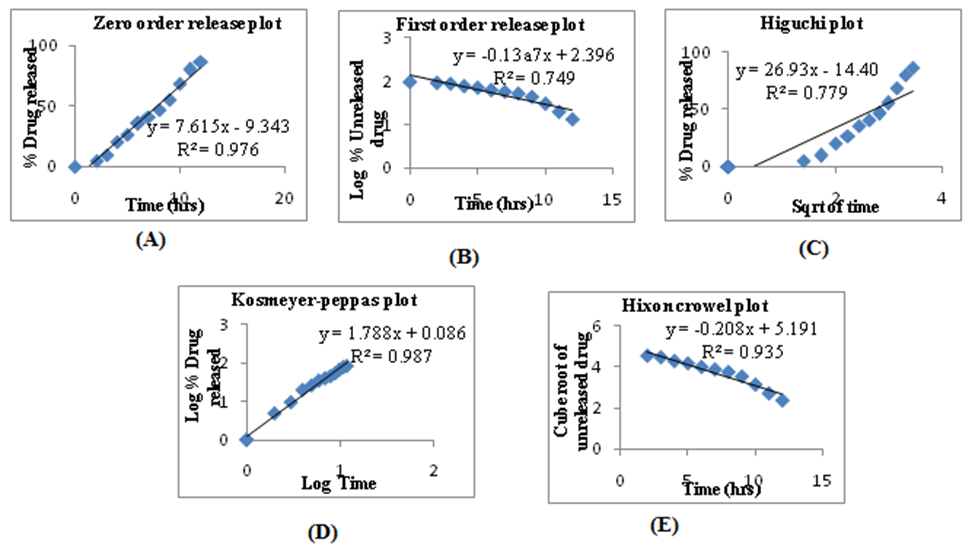
Fig. 8: Data of release kinetics (A) Zero order plot; (B) First order plot; (C) Higuchi plot; (D) Korsemeyer-Peppas plot; (E) Hixson Crowell plot
CONCLUSION
The release profile of optimized tablets showed an immediate release of 99.67% of the drug for 1hr from the outer layer followed by a lag time of 4hrs. The core tablet then release slowly for a prolonged time period to show a sustained action. They showed a release of 98.97% of the drug over a period of 12hrs. Thus the designed formulation can be considered as a promising technique for preparing press coated floating osmotic drug delivery system in chronotherapeutic management of ulcer.
5. Conflict Of Interest
There are no conflicts of interest.
6. ACKNOWLEDGEMENT
The authors would like to thank Dr. P. Udaya Shankar, Principal, Maharajah’s College of Pharmacy, Vizianagaram for providing required facilities to carry out this research work.
REFERENCES


A. Lakshmi Usha*1, A. V. S. Bhavani2, HanumanthuSri Gayatri3, Sonali Padhi4, Lahari Medikonda4, Konkyana Tejaswini5, Development And Optimization of Ranitidine HCL Press Coated Osmotic Floating Tablets, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 2513-2524. https://doi.org/10.5281/zenodo.14526291
 10.5281/zenodo.14526291
10.5281/zenodo.14526291
