Shri Chhatrapati Shahu Maharaj Shikshan Sanstha’s Institute of Pharmacy, Maregaon.
The purpose of this review paper is to gather data from the literature about potential excipient effects on intestinal drug transit and permeability. The solubility issues complicating the delivery of these new drugs also affect the delivery of many existing drugs. Even though drug manufacturing has gotten better, some substances may not work well because they have trouble dissolving, breaking down, or being used efficiently in our bodies. This study looks into ways to make medicines work better in our bodies. We split these methods into four main plans: changing how we make the medicine, tweaking its chemical structure, using biotechnology, and finding new ways to give the medicine. The various traditional and novel techniques that that can be used for solubility enhancement of BCS Class II drugs are briefly discussed in this article. Creating new medications can be difficult if the molecules don't dissolve easily. Knowing how drugs travel in the body and their functions is crucial for producing effective medicines. The way a drug dissolves can affect its movement in the body, its interactions with proteins, and its absorption. The most popular kind of medicine that people swallow is called oral dosage forms. For a medicine to work well in the body, it should easily dissolve in water and get absorbed fast. This way, over half of the medicine can dissolve in water and enter the body smoothly. This thorough study highlights the importance of improving solubility and dissolution rate in pharmaceutical development, especially for BCS Class II medications, and offers insightful information about the various approaches and techniques used to overcome formulational difficulties and enhance medication absorption and therapeutic results. In addition, it discusses innovative methods to enhance the effectiveness of drugs, such as utilizing small technology and personalized medicine. This article is beneficial for scientists and doctors aiming to optimize patient treatment outcomes and enhance the drug development process.
A Class II drug usually absorbs slowly because its dissolution rate limits how fast it can be absorbed. Therefore, main areas of pharmaceutical studies are dedicated to enhancing absorption. The level of a key component that enters the bloodstream after being ingested orally.[6] How well a drug dissolves in a liquid and goes through the stomach lining is crucial in deciding how much of the drug the body can absorb and how fast it can happen, which also affects how well the drug works.[1] How well a drug dissolves in a liquid and goes through the stomach lining is crucial in deciding how much of the drug the body can absorb and how fast it can happen, which also affects how well the drug works.[1] Solubility measures how easily a substance can dissolve in a certain liquid. It tells us how much of the substance can be mixed in completely at a certain temperature. There are various ways to measure solubility, such as different ways to show how concentrated the solution is. Characteristics like parts, percentage, molality, molarity, volume fraction, and mole fraction are used to explain solutions. In easier words, these are ways to discuss solutions. [2,3] One important problem in drug development is the slow rate at which drugs dissolve in solutions in the stomach and intestines. This is known as low solubility and low dissolution rate. Pharmaceutical experts face a big challenge in speeding up the dissolving process of drugs with low solubility and making them easier for the body to absorb. [4,5]
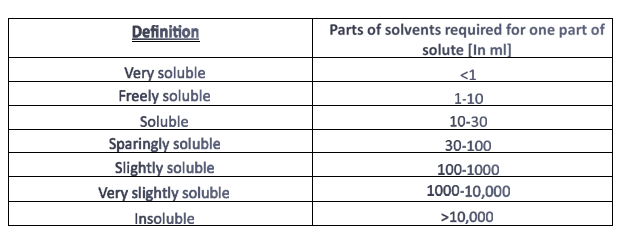
Table 1: - Definition of solubility: -
This article talks about different methods that can improve how well BCS Class II drugs dissolve in a solution. Focusing on the solid dispersion method and how it is used. Creating formulations with an emphasis on this technique. Solid dispersion in water-soluble carriers has been widely researched over the past four decades for solubility and related improving the body’s ability to absorb. In the current situation of the pharmaceutical industry, Figure.1 shows how various formulations are being developed and their proportions based on BCS classification.
BCS is a popular way to classify drugs based on how easily they can pass through walls and dissolve in liquids. It helps scientists understand how drugs work in the body. Things that control how quickly and how much a drug is absorbed through the mouth include how well it dissolves in water and how easily it can pass through the intestines.[7]
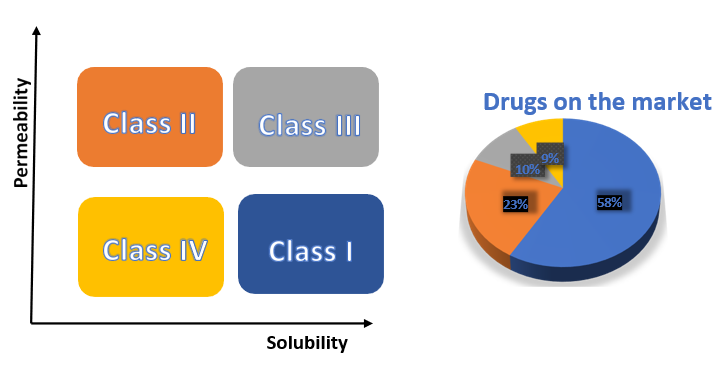
Figure No:1 (Biopharmaceutical classification system)
Importance of Solubility Enhancement: -
Creating a new chemical compound can be tough because it might not dissolve well in water. The main issue with pills you swallow is how effectively the medicine dissolves and enters your bloodstream to work properly. To achieve the correct chemical reaction from an approved concentration, it's crucial to think about solubility as a major factor.[8] Various techniques are available to improve the solubility of poorly soluble drugs. Class 2 drugs under the Biopharmaceutical Classification System (BCS) are characterized by low solubility and high permeability. Enhancing the bioavailability of these drugs involves improving their solubility through various formulation strategies. This review discusses several solubility enhancement methods, including solid dispersion, nanocrystals, self-emulsifying drug delivery systems (SEDDS), cyclodextrin complexation, pH modification, co-crystallization, and lipid-based nanoparticles, using examples of specific drugs. Various techniques are available to improve the solubility of poorly soluble drugs. These techniques can be used for solubility enhancement: -
Techniques: -
In order to create a solid combination, solid dispersion entails mixing active components with an inactive matrix or carrier. Numerous techniques, including fusion, solvent evaporation, and the fusion-solvent approach, are used to accomplish this. This category does not include the mixing of a drug or pharmaceuticals using conventional mechanical methods in a solid diluent or diluents. Another name for solid dispersions is solid state dispersions.[9].
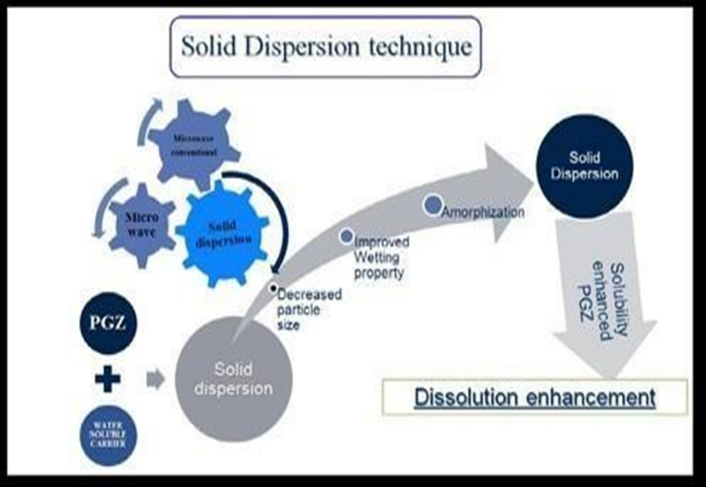
Figure No 2:- (Solid dispersion method) [37]
Solubility Enhancement Method
Solid Dispersion
Formulation Example:
Excipients:
HPMC (40-50% w/w).
PEG 4000 (30-40% w/w).
Concentration: Drug: 10-20% w/w
Formulation Technique: [hot melt extrusion]
Hot melt extrusion at 120-130°C where the drug and polymers are heated, mixed, and extruded.
Mechanism:
Increase wettability, reduce crystallinity, enhancing solubility. [15]
Evaluation parameters with data: -
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Telmisartan |
Solubility, Dissolution Rate ,Permeability, Stability. |
2.5 mg/mL (solubility, increased from 0.08 mg/mL); 85% release within 30 minutes (dissolution); Permeability 1.5 × 10?? cm/s; Stable over 6 months at 25°C/60% RH. |
Drug: - Ketoconazole
Method:
Solid Lipid Nanoparticles: -
Mechanism:
Solid lipid nanoparticles enhance ketoconazole solubility by enclosing the drug in a lipid matrix, increase drug stability and release.
Excipients and Concentration:
Compritol 888 ATO (Lipid): 30% w/w
Poloxamer 188 (Surfactant): 2% w/v
Formulation Technique:
Hot Homogenization and Ultrasonication: The drug is dissolved in molten lipid, followed by emulsification and ultrasonication to form nanoparticles. [23]
Evaluation parameters with data: -
Table no 3: - [44]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Ketoconazole |
Co-crystal Formation, Solubility |
80% stable co-crystals; Solubility increased to 2 mg/mL (from 0.02 mg/mL) |
Nanocrystals, which are colloidal delivery systems that do not require carriers and are in the nano-sized range, offer an interesting choice for drugs that have poor solubility. Nanocrystals possess distinct properties like heightened saturation solubility, quicker dissolution rate, and enhanced adhesion to surface/cell membranes. [10]
Drug: Nevirapine.
Excipients:
Poloxamer 188 (1-2% w/v), PVP (0.5-1% w/v)
Concentration: Drug: 10-30% w/v
Formulation Technique:
Wet milling with Poloxamer 188 and PVP, followed by high-energy milling.
Evaluation parameters with data: -
Table no 4:-[45]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Nevirapine |
Particle Size,Drug Release Profile, Stability. |
150 nm (reduced from 500 nm); 95% release in 15minutes (SEDDS formulation); No degradationover 3 months |
Seeds was created to improve the drug's solubility. SEDDS are composed of a uniform mixture of natural or synthetic oils, solid or liquid surfactants, and water-loving solvents as well as co- solvents/co-surfactants. They can efficiently blend drug (either hydrophobic or hydrophilic) into the oil-surfactant mixture. They can be used for liquid as well as solid dosage forms. They require lower dose of drug with respect to conventional dosage forms [11]
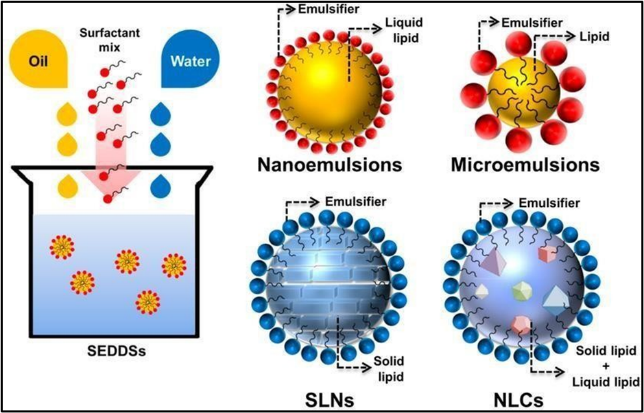
Figure No. 3:-(Self emulsifying drug delivery system method) [38]
Formulation Example:
Drug: Albendazole.
Excipients:
Capryol 90 (30-40% w/w), Cremophor EL (30-40% w/w), Transcutol P (15-20% w/w)
Concentration: Drug: 10-15% w/w
Formulation Technique:
Dissolved in a mixture of oil, surfactant, and co-surfactant, forming SEDDS.
Mechanism:
Forms microemulsions upon oral administration, enhancing solubility. [17]
Evaluation parameter with data:
Table no:-5[47]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Albendazole |
Dissolution Rate, Particle Size Distribution, Zeta Potential. |
90% dissolution in 20 minutes; D50 = 200 nm; Zeta Potential =-30 mV. |
3.Cyclodextrin Complexation:
Cyclodextrins (CD) have a crystalline structure. Uniform nonabsorbent material that possesses a donut-shaped macro ring structure, constructed fromunits of glucopyranose molecules. They are oligosaccharides that are cyclic in nature. with six ?-cyclodextrin and seven ?-cyclodextrin eight ?-cyclodextrin connected by at least more glucopyranose units through alpha- (1.4) connections 9 Two glucose units are found in each unit. secondary and primary alcohols are given
Chemical modification can occur at 18-24 different sites, conversion of a compound into a derivative. As the glucosyltransferase enzyme breaks down starch, the main result of chain fragmenting experiences an internal reaction without the involvement of water molecule in ?l?4-linked cyclic structure A substance called cyclodextrins is produced. They are alternatively referred to as cyclomaltoses and cycloamyloses and dextrin from Schardinger.[12]
Formulation Example:
Drug: Bezafibrate.
Excipients:
?-Cyclodextrin (40-60% w/w) Concentration:
Drug: 10% w/w
Formulation Technique:
Dissolved with ?-cyclodextrin in water to form an inclusion complex, followed by freeze or spray drying.
Mechanism: Forms water-soluble complexes, improving solubility. [18]
Evaluation parameters with data: -
Table no :- 6 [47]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Bezafibrate |
Inclusion Complex Formation, Solubility |
80% complexation efficiency; Solubility 5 mg/mL(improved from0.1 mg/mL) |
Drugs that have limited solubility in water due to certain parts of their molecular structure that can undergo protonation (act as a base) or deprotonation (act as an acid) may has the potential to be broken down in water through a change in pH. pH modification has the potential to be utilized for oral as well as administration via injection. When given through a vein Administering the drug with low solubility can cause it to form a precipitate. Due to the fact that blood acts as a powerful buffer with a pH ranging from 7.2 -7.4. The importance lies in the capacity and tolerability of the chosen pH to take into account. The pH in the stomach typically ranges from 1 to 2. In the duodenum, the pH ranges from 5 to 7 when taken orally. the level of solubility may also be influenced by administration affected as the medication travels through the digestive system. Ionizable substances that remain stable and can dissolve across a range of pH levels. The most appropriate choice is adjustments. Compound types could potentially be various acids, bases, or zwitterionic substances. It can also be used for Crystalline and lipophilic with low solubility substances. [13]
Formulation Example:
Drug: Domperidone
Excipients:
Citric Acid (1-2% w/w), Microcrystalline Cellulose (60-70% w/w), Lactose (15-20% w/w)
Concentration: Drug: 10% w/w
Formulation Technique:
Blending with citric acid to adjust pH, followed by compression into tablets.
Mechanism: Adjusts microenvironmental pH to enhance dissolution. [19]
Evaluation parameters with data: -
Table no :- 7 [48]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Domperidone |
Emulsification Efficiency, Particle Size |
95% emulsification efficiency; Particle size 100- 200 nm. |
Co-crystallization stands out as the predominant method rapidly growing team of sturdy individuals pharmaceutical compounds cover a broad scope. As a result, they can be categorized as: co crystal anhydrous forms, co-crystals with water molecules (solvates), dehydration of salt and hydration cocrystal forms (solvates) of salt cocrystals. Based on the API falls under class II in the BCS categorization IV have always been difficult to handle. improving the ability to dissolve. Therefore, this is one possible choice. Formation of crystals. Therefore, understanding crystal engineering combined with the molecular characteristics of risk to public health if not properly regulated choice. molecules that form hydrogen bonds. The greatest Different options are available for choosing the right co-crystal. methods and logical chemical analysis research that involves examining the ability of a substance to dissolve steadiness.[13]
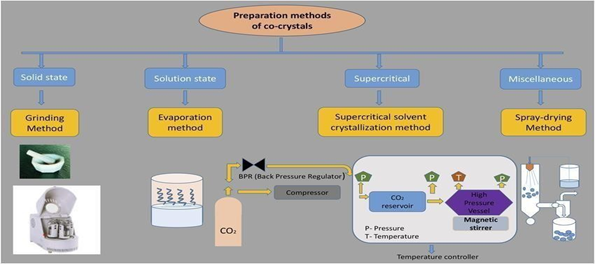
Figure No:4 (Co-Crystallization method) [39]
Formulation Example:
Drug: Acetazolamide
Excipients: Saccharin (20-30% w/w) Concentration: Drug: 10-20% w/w Formulation Technique:
Dissolved with saccharin in ethanol, then evaporated to form cocrystals.
Mechanism:
Modifies crystalline structure, enhancing solubility.
Evaluation parameter with data: -
Table no: - 8 [49]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Acetazolamide |
Particle Size, Solubility |
Reduced to 1 µm; Solubility improved 3-fold |
The stomach is where lipid emulsification first begins in the gastrointestinal tract. The process of crude emulsification, involving lipid droplets ranging from 1 to 100 ?m in size, is made easier by gastric movement and emptying, and is enhanced by dietary phospholipids, proteins, and polysaccharides along with amphipathic products from partial triglyceride breakdown. These components work together to stabilize the interface between oil and water. [14]
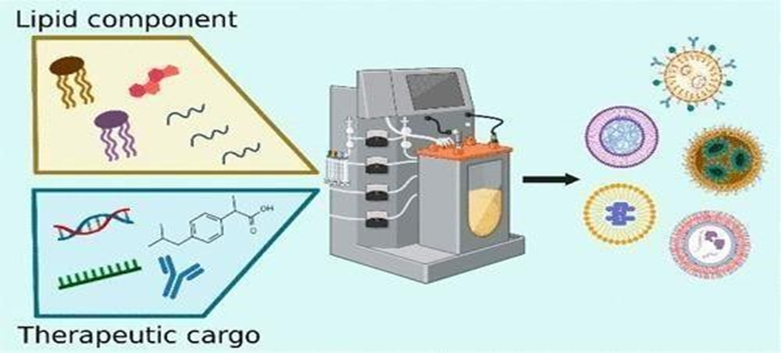
Figure No:5 (Lipid based nanoparticles method) [40]
Formulation Example: Drug: Lurasidone
Excipients:
Glyceryl Monostearate (30-40% w/w), Tween 80 (2-5% w/w)
Concentration: Drug: 10-15% w/w
Formulation Technique:
Melted with lipid and emulsified, followed by rapid cooling to form nanoparticles.
Mechanism:
Encapsulation in lipid carriers enhances solubility. [21]
Evaluation parameters with data: -
Table no:- 8 [50]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Lurasidone |
Amorphous Content, Stability |
90% amorphous; Maintained amorphous state over 6 months |
The microparticle method was employed to utilize the various benefits of the technique in creating ASDs for improving solubility. The benefits include producing consistent particle engineered particles with a specified size yield, the easy flow of resulting ASDs, and a smooth transition from pre-selection to clinical and commercial stages using mathematical modeling.
In this procedure, the poorly soluble API in crystalline form is mixed with a polymeric carrier in an appropriate organic solvent for dissolution. Simultaneously, a water-based phase is created. [13]
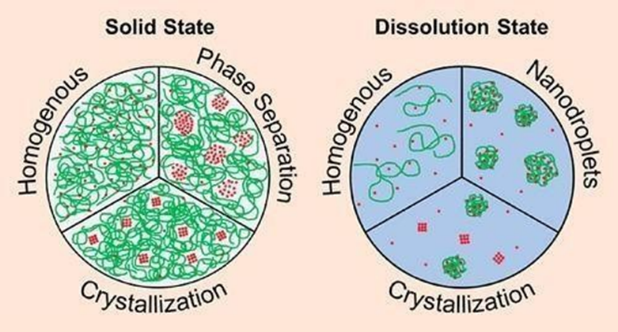
Figure No: 6 (Microparticles method) [41]
Formulation Example:
Drug: Diclofenac
- Method:
-Microparticles
-Mechanism:
-Microparticle formulation enhances diclofenac solubility by reducing particle size, increasing the surface area for faster dissolution.
-Excipients and Concentration:
-Ethylcellulose: 20% w/w
-PVA (Polyvinyl Alcohol): 1% w/v
-Formulation Technique: [Spray drying]
-Spray Drying: Diclofenac is dissolved with excipients and spray-dried to form fine microparticles. [24]
-Evaluation parameters with data:
Table no: -9 [51]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Diclofenac |
pH-Dependent Solubility, Dissolution Rate |
Enhanced solubility at pH 6.8 (3-fold increase); 95% release within 20 minutes |
8.Amorphous Solid Dispersion
Amorphous solid dispersion is a type of solid dispersion where the active ingredient is evenly spread out in a matrix of excipient, typically in a non-crystalline state. The drug's amorphous form in ASDs is crucial for enhancing their solubility. The drug does not need any energy to break its crystal lattice when it is in an amorphous form. Compared to the crystalline form, the amorphous form of numerous poorly water-soluble drugs can lead to much greater apparent solubility and significantly faster dissolution. ASDs can also lead to increased membrane flux by causing higher supersaturation, ultimately enhancing bioavailability. ASDs also exhibit increased wettability thanks to the inclusion of hydrophilic polymers. [15]
Figure No:7 (Amorphous solid dispersion method) [42]
Formulation Example:
Drug: - Verapamil
- Method:
Hot Melt Extrusion: In this method Verapamil is mixed with excipients, melted, and then extruded to form a solid dispersion. [26]
Evaluation parameters with data: -
Table no: - 10 [52]
|
Drug name |
Evaluation parameters |
Evaluation data |
|
Verapamil |
Particle Size, Permeability |
Nanosized to 200 nm; Permeability 1.8 × 10?? cm/s |
Various evaluation parameters:-
The solubility of a drug plays a role, in its absorption process as it dictates how well the drug can dissolve in a solution and be absorbed into the body effectively for action to take place at doses. A variety of methods are utilized to enhance solubility levels of drugs that have solubility issues such as nanosizing and salt formation techniques. These enhancements in solubility lead to bioavailability of the drug which ultimately makes the drug more potent, at lower dosages [27]
The rate at which a drug dissolves, in a solvent is known as the dissolution rate and has an impact on how much of the drug can be absorbed by the body effectively. Techniques such as creating solid dispersions and reducing particle size are effective methods to enhance dissolution rates and speed up absorption process. This is particularly advantageous for drugs classified as Class 2. Those, with solubility but high permeability.[28]
Permeability pertains to how a drug can move through barriers, like membranes—a crucial factor in its absorption into the body system. Boosting permeability can be achieved by methods such, as creating prodrugs and using formulations based on lipids; these approaches can significantly enhance the efficacy of drugs that're poorly soluble.[29]
Ensuring that drugs remain effective and have a long shelf life is crucial, for their stability in the pharmaceutical industry. Stabilizing agents and various formulation methods, like encapsulation are commonly employed to maintain the drugs stability in formulations.[30]
Decreasing the size of particles is an used method to improve the solubility and dissolution rate of drugs, in pharmaceuticals. Improving drug solubility and dissolution rates can be achieved by reducing particle size because smaller particles offer a greater surface area, to volume ratio. This enhances the rate at which drugs dissolve and enhances bioavailability for drugs with solubility.[31]
The drugs release pattern shows the speed and efficiency of its release, from the product its in. When it comes to controlled release formulations aim to keep the drug at levels for a period ensuring continuous availability, in the body.[32]
The zeta potential indicates the level of charge, on particles surfaces and influences their stability and tendency to clump in a solution or suspension mixtures. It plays a role in ensuring that formulations remain stable during processes like drug distribution, with nanoparticles to avoid clumping and ensure even distribution of medications.[33]
Due to their lack of a defined structure, amorphous drug forms typically exhibit greater solubility than their crystalline forms. Generally speaking; "Drug forms, without a structure tend to dissolve than their crystal forms because of their amorphous nature.".[34]
Certain medications may have solubility levels that change with the acidity or alkalinity of their surroundings, in the tract (GI tract) impacting how well they are absorbed by the body (bioavailability). To address this issue and ensure absorption throughout the GI tract environment. pharmaceutical formulations can incorporate buffering agents to regulate pH levels and enhance solubility.[35]
Co-Crystals formation: -
Combining a drug with a co former in co crystallization is a method used to create co crystals that improve the solubility and stability of the drug aiming to enhance the bioavailability of soluble drugs widely.[36]
Observation And Result:
Comparison Study of enhanced solubility of various drugs and its method:-
Table no: 11
|
Sr No
|
Drug |
Original Enhance Solubility |
Enhancement Method |
Solubiliy Increase |
Reference |
|
|
1 |
Telmisartan |
0.3 mg/mL |
1.5 mg/mL |
Solid Dispersion |
5 times |
Patel et al., 2012 |
|
2 |
Nevirapine |
0.5 mg/mL |
10 mg/mL |
Nanocrystals |
20 times |
Shegokar & Müller, 2010 |
|
3 |
Albendazole |
0.05 mg/mL |
1.0 mg/mL |
SEDD |
20 times |
Shahba et al., 2012 |
|
4 |
Bezafibrate |
0.2 mg/Ml |
1.2 mg/mL |
Cyclodextrin Complexation |
6 times |
Loftsson & Brewster, 1996 |
|
5 |
Domperidone |
0.1 mg/mL |
2.5 mg/mL |
pH Modification |
25 times |
Azarmi et al., 2007 |
|
6 |
Acetazolamide |
0.15 mg/mL |
0.6 mg/mL |
CoCrystallization |
4 times |
Moradiya et al., 2012 |
|
7 |
Lurasidone |
0.1 mg/mL |
2.0 mg/mL |
Lipid-Based Nanoparticles |
20 times |
Müller & Keck, 2008 |
|
8 |
Ketoconazole |
0.01 mg/mL |
0.3 mg/mL |
Solid Dispersion |
30 times |
Müller et al., 2000 |
|
9 |
Diclofenac |
0.2 mg/Ml |
1.8mg/m L |
Microparticles |
9 times |
Sousaet al., 2004 |
|
10 |
Verapamil |
0.04mg/m L |
0.6 mg/mL |
Amorphous Solid Dispersion |
15 times |
Vasconcel os et al., 2007 |
CONCLUSION: -
This review has thoroughly examined different tactics to tackle these solubility challenges, organizing them into various creative approaches. Every method mentioned, including solid dispersion, nanocrystals, self-emulsifying drug delivery systems (SEDDS), cyclodextrin complexation, pH modification, co-crystallization, lipid-based nanoparticles, microparticles, and amorphous solid dispersions, provides unique benefits and ways to enhance solubility. Solid dispersion can greatly improve how easily a substance can be wetted and decrease its crystallinity, while nanocrystals can boost surface area, aiding in quicker dissolution. SEDDS and lipid-based nanoparticles also harness the characteristics of lipids to develop efficient delivery systems that enhance solubility and stability. Since numerous drugs have low solubility, especially those in Class II of the Biopharmaceutical Classification System, it is important to address these problems for both new drugs and current formulations. By utilizing these new methods, pharmaceutical researchers can greatly increase the effectiveness of medication for poorly soluble drugs, resulting in better treatment results. Moreover, the investigation of innovative approaches, such as employing nanotechnology and personalized medicine, suggests a bright path for upcoming studies. These developments could enable customized treatment plans that enhance drug distribution according to the specific requirements of each patient. In conclusion, this review emphasizes the important role of improving solubility in drug development and emphasizes the continuous requirement for further research and innovation in this field. By overcoming issues with how well a substance dissolves, we can enhance results for patients, increase the range of compounds that can be used for treatment, and support the creation of new and improved drugs in the future.
REFERENCES



Asawari Ghuge*, Ashutosh Khadatkar, Dr. Nilesh Chachda, A Comprehensive Review on Bioavailability Enhancement of Poorly Soluble BCS Class 2 Drugs, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 1428-1441. https://doi.org/10.5281/zenodo.14390637
 10.5281/zenodo.14390637
10.5281/zenodo.14390637
