Faculty of Pharmaceutical Sciences, Rama University, Kanpur, India.
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia worldwide, characterized by cognitive decline, memory impairment, and neuronal loss. Despite extensive research, its pathogenesis remains complex and multifactorial, involving a network of interconnected molecular and cellular mechanisms. This review provides a comprehensive overview of the key pathological processes underlying AD, including amyloid-? (A?) accumulation, tau protein hyperphosphorylation, neuroinflammation, oxidative stress, mitochondrial dysfunction, synaptic impairment, and blood-brain barrier (BBB) disruption. While the amyloid cascade hypothesis has long dominated the field, emerging evidence highlights the critical role of tau pathology and neuroinflammatory responses in driving disease progression. Additionally, mitochondrial dysfunction and oxidative stress contribute significantly to neuronal damage, whereas vascular alterations and BBB breakdown impair A? clearance and exacerbate neurodegeneration. Importantly, these pathological pathways are highly interconnected, forming a self-perpetuating cycle that accelerates disease progression. The limited success of single-target therapeutic approaches underscores the need for multi-target and integrative strategies. This review emphasizes the importance of understanding AD as a systems-level disorder and discusses current therapeutic implications and future perspectives, including biomarker-driven diagnosis, precision medicine, and combination therapies. A deeper understanding of these interconnected mechanisms is essential for the development of effective disease-modifying interventions aimed at slowing or preventing AD progression.
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disorder characterized by a gradual decline in cognitive function, memory impairment, and behavioral disturbances. It represents the most common cause of dementia, accounting for approximately 60-70% of cases worldwide, and poses a significant and growing global health burden due to increased life expectancy and aging populations as per WHO, 2023 [1]. Despite decades of research, the precise etiology of AD remains elusive, reflecting its multifactorial and complex nature involving a convergence of genetic, molecular, and environmental factors. The classical pathological hallmarks of AD include extracellular deposition of amyloid-β (Aβ) plaques and intracellular accumulation of hyperphosphorylated tau protein forming neurofibrillary tangles (NFTs). The amyloid cascade hypothesis, long considered the central framework of AD pathogenesis, posits that aberrant production and aggregation of Aβ peptides initiate a cascade of neurotoxic events leading to synaptic dysfunction and neuronal loss [2]. However, increasing evidence suggests that amyloid pathology alone is insufficient to fully explain disease progression, shifting attention toward a more integrative understanding of disease mechanisms.
Tau pathology has emerged as a critical determinant of neurodegeneration, with abnormal tau phosphorylation, aggregation, and propagation correlating more closely with cognitive decline than amyloid burden [3]. The spread of pathological tau across interconnected brain regions highlights the prion-like behavior of tau aggregates and underscores its role in disease progression. Additionally, synaptic dysfunction and loss, which occur early in AD, are now recognized as key contributors to cognitive impairment, further emphasizing the importance of cellular-level alterations in disease onset. Beyond protein aggregation, neuroinflammation plays a pivotal role in AD pathogenesis. Activated microglia and astrocytes contribute to a chronic inflammatory environment, releasing cytokines and reactive oxygen species (ROS) that exacerbate neuronal injury [4]. Genome-wide association studies (GWAS) have identified several immune related genes, such as TREM2 and CD33, implicating innate immune dysregulation as a central component of AD pathology [5].Mitochondrial dysfunction and oxidative stress further contribute to neuronal damage by impairing energy metabolism and increasing the production of free radicals. Neurons, being highly energy-dependent, are particularly vulnerable to mitochondrial abnormalities, which have been consistently observed in AD brains [6]. In parallel, impaired proteostasis mechanisms, including dysfunction of the ubiquitin-proteasome system and autophagy-lysosomal pathways, lead to the accumulation of toxic protein aggregates, thereby accelerating neurodegeneration. Recent advances have also highlighted the role of vascular contributions and blood-brain barrier (BBB) dysfunction in AD. Cerebrovascular alterations, reduced cerebral blood flow, and BBB breakdown facilitate the entry of peripheral inflammatory mediators into the brain, amplifying neuroinflammatory responses and promoting disease progression [7]. Furthermore, metabolic dysregulation, including insulin resistance and altered lipid metabolism, has been implicated in AD, leading to the concept of type 3 diabetes as a contributing factor. Importantly, AD is increasingly viewed as a disorder of interconnected pathological pathways rather than a single-cause disease. The interplay between amyloid deposition, tau pathology, neuroinflammation, oxidative stress, mitochondrial dysfunction, and vascular impairment creates a self-perpetuating cycle that drives progressive neuronal damage. This paradigm shift has significant implications for therapeutic strategies, emphasizing the need for multi-targeted approaches rather than single-pathway interventions.
2. Methodology
This review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA 2020) guidelines to ensure a systematic and transparent approach. A comprehensive literature search was performed across PubMed, Scopus, Web of Science, and Google Scholar databases for studies published between January 2022 and December 2025. Relevant articles were identified using a combination of keywords including "Alzheimer's disease," "pathogenesis," "amyloid beta," "tau protein," "neuroinflammation," "oxidative stress," and "mitochondrial dysfunction." Only peer-reviewed articles published in English and focusing on molecular and cellular mechanisms of AD were included, while editorials, conference abstracts, and non-relevant studies were excluded. After removal of duplicates, studies were screened based on titles and abstracts, followed by full-text assessment according to predefined eligibility criteria. Data were systematically extracted and qualitatively synthesized into key pathological domains to provide an integrated understanding of AD pathogenesis.
3. Molecular and Cellular Mechanism Underlaying AD Pathogenesis
3.1. Amyloid-β Pathology
Amyloid-β (Aβ) pathology is widely recognized as a central event in the pathogenesis of AD, characterized by the abnormal production, aggregation, and deposition of Aβ peptides in the brain. Aβ is generated through the sequential proteolytic cleavage of amyloid precursor protein (APP) by β-secretase (BACE1) and y-secretase, resulting primarily in Aβ-40 and the more aggregation-prone Aβ-42 isoforms. Among these, Aβ 42 exhibits a higher tendency to form oligomers and fibrils, which are considered the most neurotoxic species [2, 8]. The amyloid cascade hypothesis proposes that the accumulation of Aβ initiates a series of downstream pathological events, including synaptic dysfunction, tau hyperphosphorylation, neuroinflammation, and ultimately neuronal death. Soluble Aβ oligomers, rather than insoluble plaques, are now regarded as the primary mediators of synaptic toxicity, impairing long-term potentiation (LTP) and disrupting neurotransmission, particularly in hippocampal circuits critical for memory formation [9]. These oligomers interfere with synaptic receptors such as NMDA and AMPA receptors, leading to calcium dysregulation and excitotoxicity.In addition to synaptic impairment, Aβ aggregation induces oxidative stress by generating reactive oxygen species (ROS), which damage lipids, proteins, and nucleic acids. Mitochondrial dysfunction is closely associated with Aβ toxicity, as Aβ can localize within mitochondria and disrupt electron transport chain activity, leading to reduced ATP production and increased oxidative burden [6]. This metabolic impairment further exacerbates neuronal vulnerability and accelerates neurodegeneration (figure 1).Aβ also plays a significant role in activating neuroinflammatory pathways. Aggregated Aβ interacts with microglial receptors such as TREM2, triggering microglial activation and the release of pro-inflammatory cytokines. While microglia initially attempt to clear Aβ deposits through phagocytosis, chronic activation results in a sustained inflammatory response that contributes to neuronal damage [10]. Genetic studies have further reinforced the importance of Aβ in AD, with mutations in APP, PSEN1, and PSEN2 genes leading to increased Aβ production and early-onset familial AD [11]. Recent evidence suggests that impaired clearance mechanisms, rather than overproduction alone, significantly contribute to Aβ accumulation in sporadic AD. Dysfunction of the glymphatic system, reduced enzymatic degradation (e.g., neprilysin and insulin-degrading enzyme), and compromised blood-brain barrier (BBB) transport collectively hinder Aβ clearance from the brain [12]. Furthermore, vascular dysfunction and reduced cerebral perfusion exacerbate Aβ deposition, linking amyloid pathology with cerebrovascular abnormalities. Despite its central role, the amyloid-centric view of AD has been increasingly challenged, as clinical trials targeting Aβ have yielded limited success in halting disease progression. This has led to a paradigm shift toward understanding Aβ as one component of a multifactorial network of pathological processes rather than the sole driver of disease. Nonetheless, Aβ pathology remains a critical upstream event that interacts with other molecular and cellular mechanisms to drive the complex progression of AD.
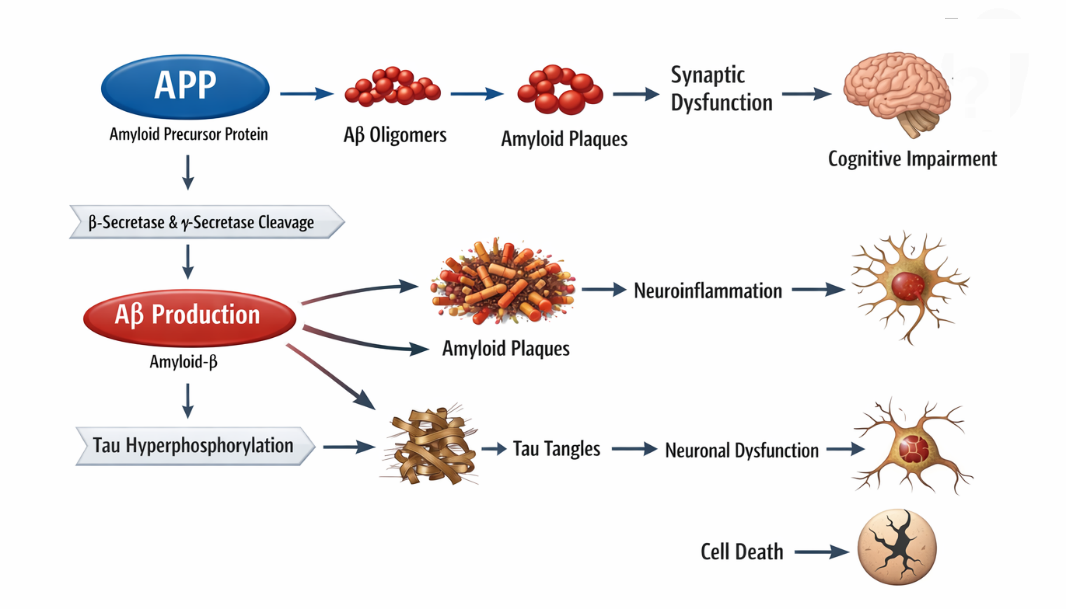
Figure 1. Schematic representation of amyloid-β (Aβ)-mediated pathogenic events in Alzheimer’s disease
3.2. Tau Protein Dysfunction
Tau pathology represents a critical and closely correlated determinant of neurodegeneration in AD, often showing a stronger association with cognitive decline than Aβ burden. Tau is a microtubule-associated protein primarily expressed in neurons, where it stabilizes microtubules and supports axonal transport. In AD, tau undergoes abnormal post-translational modifications, particularly hyperphosphorylation, which reduces its affinity for microtubules and promotes its aggregation into paired helical filaments and neurofibrillary tangles (NFTs) [3]. The detachment of hyperphosphorylated tau from microtubules leads to cytoskeletal destabilization and impaired axonal transport, disrupting the delivery of essential organelles and nutrients to synapses. This contributes significantly to synaptic dysfunction and neuronal degeneration. Moreover, tau aggregates interfere with intracellular signaling pathways and promote neuronal toxicity through gain-of-function mechanisms [13].
A key feature of tau pathology is its prion-like propagation across interconnected brain regions. Misfolded tau can spread trans-synaptically from one neuron to another, seeding further aggregation and facilitating the progression of pathology in a stereotypical pattern, as described by Braak staging. This spatial and temporal spread of tau pathology correlates strongly with disease severity and clinical symptoms [14].
Emerging evidence also highlights the interaction between amyloid-? and tau pathology. A? accumulation is thought to act upstream, triggering tau hyperphosphorylation through kinase activation (e.g., GSK-3β and CDK5), thereby linking the two hallmark pathologies of AD. This synergistic interaction accelerates neurodegeneration and amplifies disease progression [15]. In addition to phosphorylation, other post-translational modifications such as acetylation, truncation, and ubiquitination further influence tau aggregation and toxicity. Impairment of proteostasis mechanisms, including autophagy and the ubiquitin-proteasome system, contributes to the accumulation of pathological tau species within neurons. Furthermore, tau pathology has been associated with mitochondrial dysfunction and increased oxidative stress, reinforcing its role in cellular damage (figure 2). Given its strong correlation with cognitive impairment and disease progression, tau has emerged as a promising therapeutic target. Strategies aimed at inhibiting tau aggregation, enhancing its clearance, or preventing its propagation are currently under active investigation. These approaches underscore the importance of tau not only as a pathological hallmark but also as a central mediator of neurodegeneration in AD.
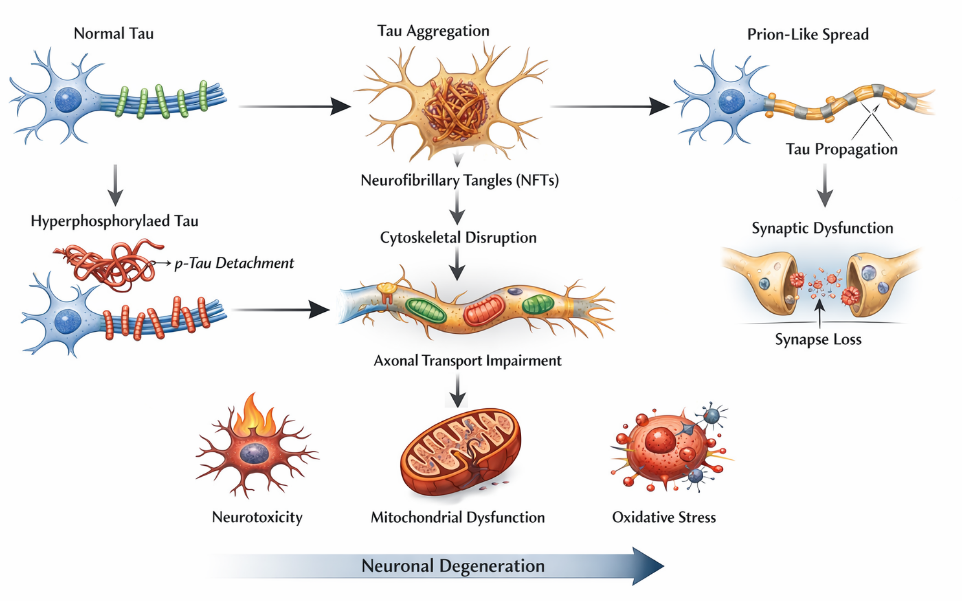
Figure 2. Schematic representation of tau protein dysfunction in Alzheimer’s disease pathology
3.3. Neuroinflammation
Neuroinflammation has emerged as a central and dynamic contributor to the pathogenesis of AD, playing a dual role in both protective and detrimental processes within the brain. It is primarily mediated by glial cells, particularly microglia and astrocytes, which respond to pathological stimuli such as Aβ accumulation and tau aggregation. While acute activation of these cells may initially facilitate the clearance of toxic proteins, chronic and dysregulated neuroinflammation leads to sustained neuronal damage and accelerates disease progression [4, 16, 17]. Microglia, the resident immune cells of the central nervous system, are among the first responders to Aβ deposition. Upon activation, microglia attempt to phagocytose Aβ plaques; however, prolonged exposure results in a shift toward a pro-inflammatory phenotype. This activated state is characterized by the release of cytokines such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6), as well as reactive oxygen and nitrogen species, all of which contribute to neuronal injury and synaptic dysfunction [18-20]. Additionally, chronic microglial activation impairs their phagocytic capacity, reducing their ability to clear Aβ and thereby exacerbating its accumulation.
Genetic studies have strongly implicated immune-related pathways in AD, highlighting the importance of microglial function. Variants in genes such as TREM2, CD33, and CR7 have been associated with altered immune responses and increased AD risk. In particular, TREM2 plays a crucial role in microglial activation, survival, and phagocytosis, and its dysfunction has been linked to impaired clearance of amyloid plaques and enhanced neuroinflammation [11, 21]. Astrocytes also contribute significantly to neuroinflammation in AD. Reactive astrocytes undergo morphological and functional changes, leading to the release of inflammatory mediators and disruption of neuronal support functions, including neurotransmitter regulation and maintenance of the BBB. Furthermore, astrocytes can amplify inflammatory signaling by interacting with activated microglia, creating a self-perpetuating cycle of inflammation [22-24].
Another critical component of neuroinflammation in AD is the activation of intracellular signaling pathways such as the NLRP3 inflammasome, which is triggered by Aß and tau aggregates. Activation of NLRP3 leads to the maturation and release of pro-inflammatory cytokines, particularly IL-1B, thereby intensifying inflammatory responses and promoting neurodegeneration [25, 26]. This pathway represents a key link between protein aggregation and immune activation in AD (figure 3). Neuroinflammation is also closely associated with oxidative stress and mitochondrial dysfunction, as activated glial cells produce excessive ROS, further damaging neuronal structures. Additionally, chronic inflammation contributes to BBB disruption, allowing peripheral immune cells and inflammatory mediators to infiltrate the brain, thereby amplifying neurodegenerative processes [27-29]. The transition from a protective to a harmful inflammatory response is a critical determinant of disease progression. Consequently, targeting neuroinflammatory pathways has gained significant attention as a therapeutic strategy, with approaches aimed at modulating microglial activation, inhibiting inflammasome signaling, and restoring immune homeostasis.
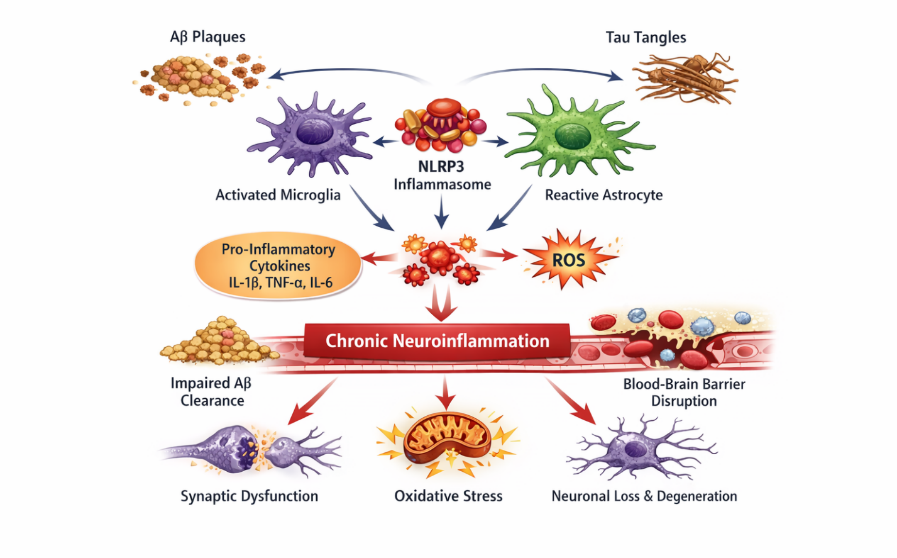
Figure 3. Neuroinflammatory mechanisms contributing to AD pathogenesis
3.4. Oxidative stress and Mitochondrial Dysfunction
Oxidative stress and mitochondrial dysfunction are central contributors to the pathogenesis of AD, playing a crucial role in neuronal damage and disease progression. The brain is particularly vulnerable to oxidative injury due to its high oxygen consumption, abundant lipid content, and relatively low antioxidant defenses. In AD, an imbalance between the production of ROS and the antioxidant defense system leads to oxidative stress, resulting in damage to cellular macromolecules, including lipids, proteins, and DNA [13, 30, 31]. Mitochondria are the primary source of ROS in neurons and are essential for maintaining cellular energy homeostasis. In AD, mitochondrial dysfunction is characterized by impaired electron transport chain activity, decreased ATP production, altered mitochondrial dynamics, and increased ROS generation. Aβ has been shown to localize within mitochondria, where it disrupts key enzymatic processes and enhances oxidative damage. Similarly, pathological tau impairs mitochondrial transport and function, further exacerbating energy deficits and neuronal vulnerability [32-34].
Oxidative stress also promotes lipid peroxidation, leading to the formation of toxic byproducts such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), which disrupt membrane integrity and impair neuronal signaling. Protein oxidation and DNA damage further compromise cellular function, ultimately triggering apoptotic pathways. Additionally, oxidative stress amplifies other pathological processes, including Aβ aggregation and tau hyperphosphorylation, creating a vicious cycle that accelerates neurodegeneration [35, 36].
Mitochondrial dynamics, including fission and fusion processes, are also disrupted in AD. Excessive mitochondrial fragmentation and impaired mitophagy lead to the accumulation of damaged mitochondria, further increasing oxidative burden. Defective mitochondrial biogenesis and altered calcium homeostasis contribute to synaptic dysfunction and neuronal loss [37, 38]. Furthermore, oxidative stress is closely linked with neuroinflammation, as activated microglia produce large amounts of ROS and reactive nitrogen species (RNS), intensifying neuronal injury. The interplay between oxidative stress, mitochondrial dysfunction, amyloid pathology, and tau abnormalities highlights the integrated nature of AD pathogenesis (figure 4). Given its pivotal role, targeting oxidative stress and mitochondrial dysfunction has emerged as a promising therapeutic strategy. Antioxidants, mitochondrial protectants, and agents that enhance mitophagy and bioenergetic function are being actively explored to mitigate neuronal damage and slow disease progression.
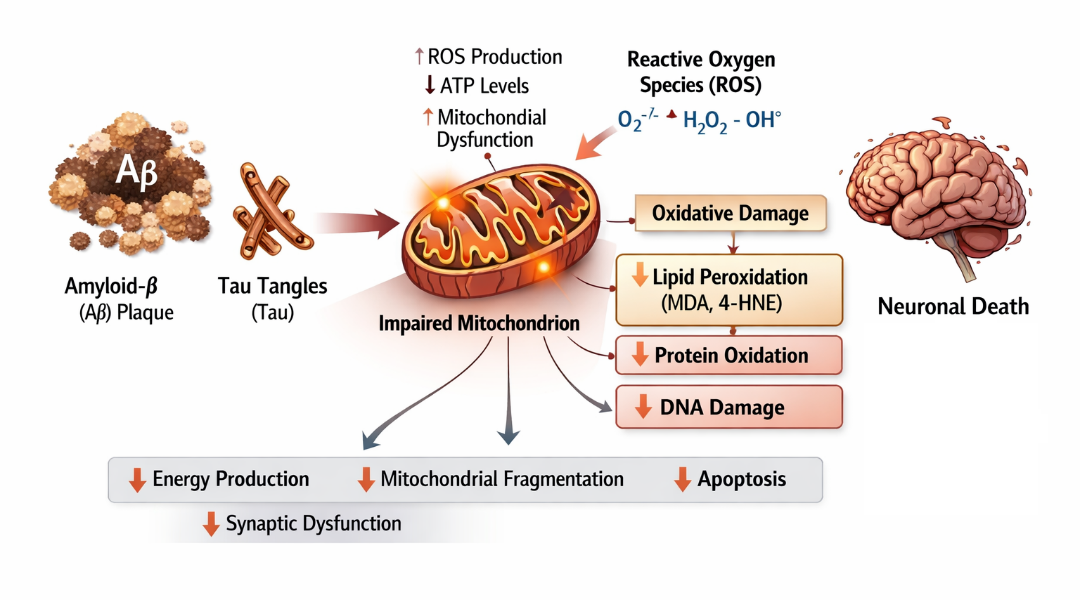
Figure 4. Schematic illustration depicting the role of oxidative stress and mitochondrial impairment in AD pathogenesis.
3.5. Synaptic Dysfunction & Neuronal Loss
Synaptic dysfunction and neuronal loss represent the ultimate pathological consequences of AD and are the primary correlates of cognitive decline and memory impairment. Among all pathological features, synaptic loss shows the strongest correlation with disease severity, emphasizing its critical role in the progression of AD [9, 39-41]. Synapses are essential for neuronal communication, plasticity, and memory formation. In AD, early synaptic impairment is primarily driven by soluble Aβ oligomers, which disrupt synaptic signaling and plasticity. Aβ oligomers interfere with key synaptic receptors, including NMDA and AMPA receptors, leading to impaired long-term potentiation (LTP) and enhanced long-term depression (LTD), thereby weakening synaptic strength and connectivity [42, 43]. This synaptic toxicity occurs even before the formation of visible amyloid plaques, highlighting its role as an early event in disease pathogenesis.
Tau pathology further exacerbates synaptic dysfunction by disrupting cytoskeletal integrity and impairing axonal transport. Mislocalized tau accumulates in dendritic spines, where it interferes with synaptic signaling pathways and contributes to synapse degeneration. The combined effects of Aβ and tau create a synergistic disruption of neuronal communication, accelerating cognitive decline [44-46]. In addition to protein aggregation, neuroinflammation significantly contributes to synaptic damage. Activated microglia can aberrantly prune synapses through complement-mediated pathways, particularly involving proteins such as C1q and C3. While synaptic pruning is a normal physiological process during development, its dysregulation in AD leads to excessive synapse elimination and functional impairment [47].
Neuronal loss in AD is a progressive process resulting from cumulative cellular stress, including oxidative damage, mitochondrial dysfunction, excitatoxicity, and chronic inflammation. Apoptotic pathways are activated in response to these stressors, leading to programmed cell death. Excitotoxicity, driven by excessive glutamate signaling and calcium influx, further contributes to neuronal injury and degeneration. Brain regions such as the hippocampus and cerebral cortex are particularly vulnerable to neuronal loss, which explains the characteristic deficits in memory, learning, and executive function observed in AD patients (figure 5). As neuronal networks deteriorate, the brain's ability to compensate diminishes, leading to irreversible cognitive decline [48-50]. Importantly, synaptic dysfunction precedes overt neuronal loss, making it a critical target for early therapeutic intervention. Strategies aimed at preserving synaptic integrity, modulating excitotoxicity, and preventing abnormal protein interactions are being actively explored to slow disease progression.
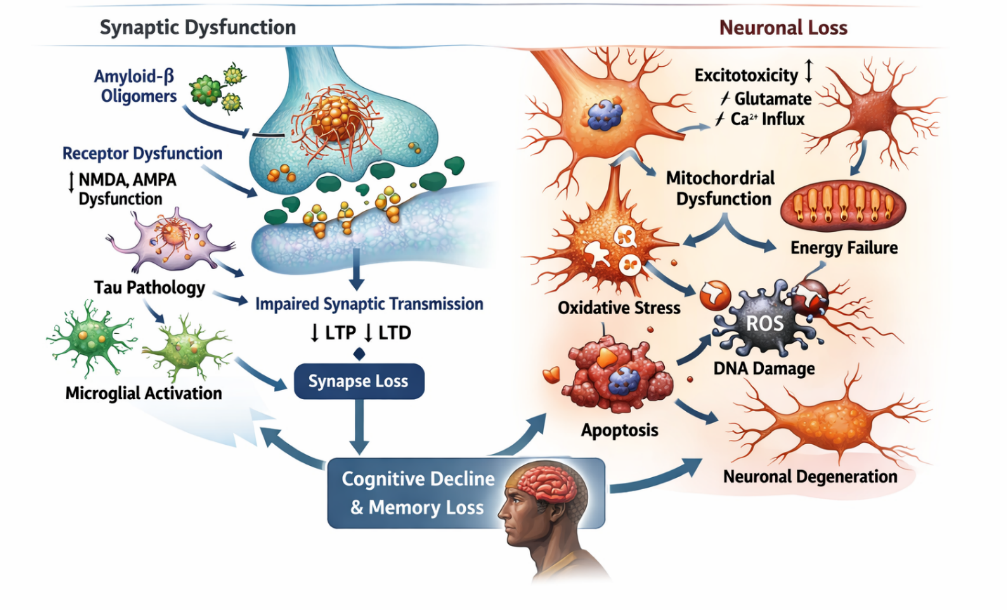
Figure 5. Synaptic dysfunction and neuronal loss in AD pathogenesis
3.6. Blood-Brain Barrier Dysfunction and Vascular Contributions
Blood-brain barrier (BBB) dysfunction and cerebrovascular abnormalities have emerged as critical components in the pathogenesis of AD, contributing significantly to disease onset and progression. The BBB is a highly selective and dynamic interface composed of endothelial cells, pericytes, astrocytic end-feet, and tight junction proteins, which collectively regulate the transport of molecules between the bloodstream and the brain. In AD, disruption of BBB integrity leads to impaired homeostasis, facilitating the entry of neurotoxic substances and peripheral immune cells into the central nervous system [12, 51, 52]. One of the key consequences of BBB dysfunction is the impaired clearance of Aβ from the brain. Under normal conditions, Aβ is transported across the BBB via receptor-mediated mechanisms involving low-density lipoprotein receptor-related protein 1 (LRP1). However, in AD, reduced expression of LRP1 and increased activity of receptors such as RAGE (receptor for advanced glycation end products) promote Aβ accumulation within the brain parenchyma, thereby accelerating plaque formation [53, 54]. Cerebrovascular dysfunction, including reduced cerebral blood flow (CBF), endothelial damage, and microvascular degeneration, further exacerbates neuronal injury. Chronic hypoperfusion leads to insufficient oxygen and glucose supply, resulting in metabolic stress and promoting oxidative damage. These vascular alterations are particularly detrimental to highly energy-dependent brain regions such as the hippocampus, thereby contributing to cognitive impairment [17, 55].
Pericyte loss is another critical factor associated with BBB breakdown in AD. Pericytes play a vital role in maintaining vascular stability and regulating capillary blood flow. Their degeneration leads to increased BBB permeability, reduced clearance of toxic metabolites, and enhanced neuroinflammatory responses. This creates a feedback loop in which vascular dysfunction and neuroinflammation mutually reinforce each other, accelerating neurodegeneration [56, 57].
BBB disruption also facilitates the infiltration of peripheral immune cells and inflammatory mediators into the brain, amplifying neuroinflammatory processes. This increased permeability contributes to the accumulation of cytokines, chemokines, and plasma proteins, which further impair neuronal function and synaptic integrity. In addition [16, 58], vascular risk factors such as hypertension, diabetes, and atherosclerosis have been strongly linked to AD, Highlighting the role of systemic vascular health in disease development. These conditions contribute to endothelial dysfunction, oxidative stress, and chronic inflammation, thereby increasing susceptibility to neurodegeneration. Importantly, BBB dysfunction is considered an early event in AD pathogenesis, occurring prior to significant neuronal loss (figure 6). This has shifted the focus toward the neurovascular unit as a therapeutic target. Strategies aimed at restoring BBB integrity, improving cerebral blood flow, and enhancing Aβ clearance mechanisms are being actively explored to slow disease progression.
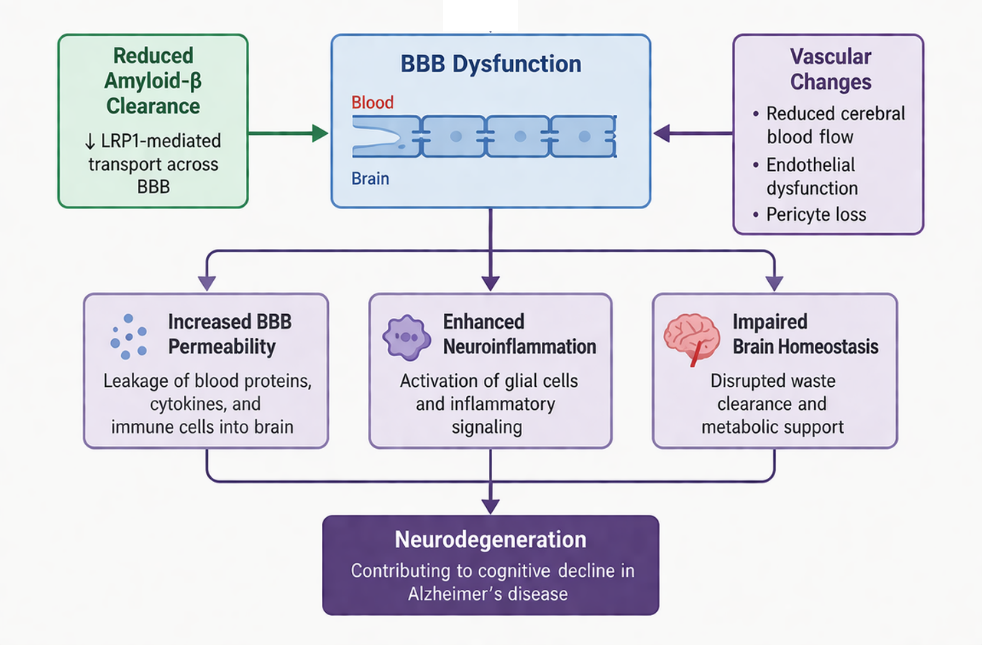
Figure 6. Blood-brain barrier dysfunction and cerebrovascular abnormalities in the pathogenesis of AD
4. Interconnection of the pathological pathways in AD
AD is increasingly recognized as a multifactorial disorder driven by the complex interplay of interconnected molecular and cellular mechanisms rather than a single pathogenic event. The classical hallmarks Aβ accumulation and tau pathology-act as central triggers that initiate and propagate a cascade of downstream processes, including neuroinflammation, oxidative stress, mitochondrial dysfunction, synaptic impairment, and vascular abnormalities. These pathways do not operate in isolation; instead, they form a highly integrated and self-amplifying network that drives progressive neurodegeneration. Aβ accumulation is widely considered an upstream event that initiates multiple pathological cascades. Soluble Aβ oligomers not only disrupt synaptic function but also activate microglia and astrocytes, thereby inducing neuroinflammatory responses. In parallel, Aβ promotes tau hyperphosphorylation through the activation of kinases such as glycogen synthase kinase- 3β (GSK-3β), linking amyloid pathology to tau-mediated neurodegeneration [15, 59-62]. Conversely, pathological tau exacerbates Aβ toxicity by impairing neuronal transport systems and facilitating synaptic dysfunction, highlighting a bidirectional relationship between these two hallmark proteins.
Neuroinflammation acts as a central hub connecting multiple pathological processes. Chronic activation of microglia leads to the release of pro-inflammatory cytokines and ROS, which contribute to oxidative stress and neuronal injury. In turn, oxidative stress enhances Aβ aggregation and tau phosphorylation, creating a vicious cycle that perpetuates cellular damage [63]. Mitochondrial dysfunction further amplifies this cycle by increasing ROS production and reducing ATP availability, thereby compromising neuronal survival and synaptic activity. Synaptic dysfunction represents a critical convergence point of these interconnected pathways. Aβ oligomers, tau aggregates, oxidative stress, and inflammatory mediators collectively impair synaptic plasticity and neurotransmission. Microglia-mediated synaptic pruning, driven by complement activation, further accelerates synapse loss. As synaptic integrity declines, neuronal networks become progressively disrupted, ultimately leading to neuronal death and cognitive impairment [64, 65].
Vascular dysfunction and BBB breakdown further integrate into this pathological network. Impaired BBB integrity reduces Aβ clearance and allows peripheral inflammatory mediators to enter the brain, thereby exacerbating neuroinflammation. Reduced cerebral blood flow contributes to hypoxia, oxidative stress, and metabolic deficits, all of which intensify neuronal vulnerability [45, 66]. Additionally, vascular risk factors such as hypertension and diabetes interact with these mechanisms, further accelerating disease progression. Importantly, these interconnected pathways create a self-perpetuating feedback loop, where each pathological process reinforces others. For example, Aβ-induced neuroinflammation increases oxidative stress, which in turn promotes further Aβ deposition and tau pathology. Similarly, mitochondrial dysfunction enhances ROS production, which exacerbates inflammation and synaptic damage. This network-based model explains why targeting a single pathway has shown limited success in clinical trials [67-69].
Therefore, AD should be viewed as a systems-level disorder, requiring multi-targeted therapeutic approaches that simultaneously address amyloid pathology, tau dysfunction, neuroinflammation, oxidative stress, and vascular impairment. Understanding the intricate interconnections among these pathways is essential for the development of effective disease-modifying therapies and for advancing precision medicine strategies in AD.
5. Therapeutic Implications
The multifactorial and interconnected nature of AD pathogenesis has profound implications for therapeutic development, highlighting the limitations of single-target strategies and the need for integrated, multi-modal approaches. Historically, therapeutic efforts have predominantly focused on the Aβ cascade; however, clinical outcomes have been modest, suggesting that targeting Aβ alone may be insufficient to halt or reverse disease progression [70]. This has led to a paradigm shift toward therapies that simultaneously address multiple pathological pathways, including tau dysfunction, neuroinflammation, oxidative stress, mitochondrial impairment, synaptic loss, and vascular abnormalities. Targeting tau pathology has gained increasing attention due to its strong correlation with cognitive decline. Therapeutic strategies under investigation include inhibition of tau phosphorylation, prevention of tau aggregation, and blockade of trans-synaptic tau propagation. These approaches aim to directly reduce neurofibrillary tangle formation and limit neuronal damage [71]. In parallel, modulation of neuroinflammatory pathways has emerged as a promising avenue, with efforts focused on regulating microglial activation, inhibiting pro-inflammatory cytokine release, and targeting key signaling mechanisms such as the NLRP3 inflammasome [72,73].
Given the central role of oxidative stress and mitochondrial dysfunction, antioxidant-based therapies and mitochondrial-targeted interventions are being actively explored. Compounds that enhance mitochondrial bioenergetics, reduce ROS production, and promote mitophagy may help restore neuronal function and delay disease progression [12, 74]. These strategies are particularly relevant in early disease stages, where oxidative damage is prominent. Preservation of synaptic integrity is another critical therapeutic objective. Approaches aimed at modulating excitotoxicity, enhancing synaptic plasticity, and preventing complement-mediated synaptic pruning are under investigation. Neuroprotective agents that stabilize synaptic function may offer significant benefits in maintaining cognitive performance.
Increasing evidence also supports targeting the neurovascular unit, including restoration of BBB integrity and improvement of cerebral blood flow. Strategies that enhance Aβ clearance through BBB transporters such as LRP1, reduce vascular inflammation, and protect endothelial and pericyte function are gaining attention as potential disease-modifying interventions [75, 76]. Importantly, the concept of combination therapy is emerging as a rational and necessary approach for AD treatment. By simultaneously targeting multiple interconnected pathways, combination therapies may overcome the limitations of monotherapies and provide synergistic therapeutic effects. This approach aligns with the systems-level understanding of AD as a network-driven disorder.
6. Future Prospective
Future investigations should prioritize elucidating the dynamic interactions among Aβ, tau pathology, neuroinflammation, oxidative stress, mitochondrial dysfunction, and vascular impairment to better define disease trajectories and identify stage-specific intervention points. Advanced multi-omics approaches-including genomics, proteomics, metabolomics, and transcriptomics-combined with systems biology and artificial intelligence (AI)-driven analytics, hold significant promise in uncovering novel molecular targets and predictive biomarkers for early diagnosis and progression. A major focus of future research will be the development of reliable and minimally invasive biomarkers for early detection and monitoring. Blood-based biomarkers, such as plasma Aβ, phosphorylated tau (p-tau), and neurofilament light chain (NLC), are emerging as valuable tools for identifying preclinical AD and tracking therapeutic responses. Integration of these biomarkers with neuroimaging techniques, including PET and advanced MRI modalities, may enable more accurate patient stratification and personalized treatment approaches [77].
The concept of precision medicine is expected to play a transformative role in AD management. Given the heterogeneity of the disease, tailoring therapeutic interventions based on individual genetic profiles, biomarker signatures, and disease stage may significantly improve clinical outcomes. For example, targeting specific pathways such as neuroinflammation or mitochondrial dysfunction in selected patient populations could enhance treatment efficacy and reduce off-target effects. Emerging therapeutic strategies are increasingly focused on multi-target and combination therapies, reflecting the interconnected nature of AD pathology. Future drug development may involve rational combinations of anti-amyloid, anti-tau, anti-inflammatory, antioxidant, and neuroprotective agents to achieve synergistic effects. Additionally, novel therapeutic modalities such as gene therapy, RNA-based interventions, and immunotherapies are being explored to modulate disease mechanisms at a molecular level.
Another promising direction is the targeting of the neurovascular unit and BBB. Restoring BBB integrity, improving cerebral blood flow, and enhancing clearance of neurotoxic proteins may provide new avenues for disease modification. Furthermore, increasing attention is being given to the role of the gut-brain axis, microbiome alterations, and systemic inflammation in AD, opening new interdisciplinary research pathways. Advancements in cell-based models, such as induced pluripotent stem cells (iPSCs) and brain organoids, along with improved animal models, will further enhance our understanding of disease mechanisms and facilitate drug screening. These models provide more physiologically relevant systems to study complex cellular interactions and test therapeutic interventions.
Importantly, future research should emphasize early intervention and prevention strategies, targeting individuals at risk before the onset of significant neurodegeneration. Lifestyle modifications, including diet, physical activity, cognitive engagement, and management of vascular risk factors, are increasingly recognized as important components of AD prevention.
CONCLUSION
Alzheimer's disease (AD) is a complex and multifactorial neurodegenerative disorder driven by the intricate interplay of diverse molecular and cellular mechanisms. While Aβ accumulation and tau pathology remain the defining hallmarks, growing evidence highlights the critical contributions of neuroinflammation, oxidative stress, mitochondrial dysfunction, synaptic impairment, and BBB disruption in disease progression. These interconnected pathways form a self-amplifying network that ultimately leads to neuronal loss and cognitive decline. The limited success of single-target therapeutic strategies underscores the need to move beyond reductionist approaches and adopt a more integrative perspective of AD pathogenesis. Targeting multiple pathological processes simultaneously, particularly at early stages of the disease, may offer greater potential for effective disease modification. Advances in biomarker development, neuroimaging, and multi-omics technologies are enabling earlier diagnosis and more precise characterization of disease subtypes, thereby paving the way for personalized therapeutic interventions. Importantly, understanding AD as a systems-level disorder provides a strong rationale for the development of multi-target and combination therapies that address the complex interactions among its underlying mechanisms. In parallel, preventive strategies focusing on modifiable risk factors and early intervention hold promise in reducing disease burden.In conclusion, a comprehensive and integrative understanding of AD pathogenesis is essential for the development of effective therapeutic strategies. Continued research efforts aimed at unraveling the complex network of pathological pathways will be critical in advancing toward disease-modifying treatments and improving clinical outcomes for patients affected by this devastating disorder.
REFERENCES


Chanchal Yadav, Nalini Kanta Sahoo, A Comprehensive Review of Alzheimer's Disease Pathogenesis: Molecular and Cellular Perspectives, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 1224-1242, https://doi.org/10.5281/zenodo.19467646
 10.5281/zenodo.19467646
10.5281/zenodo.19467646
