Fortrea, North Carolina, USA
Wilson Disease is a rare metabolic disorder with genetic origins. It causes accumulation of copper in various organs, especially the liver and brain. Symptoms include liver dysfunction, neurological disorders, and psychiatric disorders. The genetic basis of WD is mutation in the ATP7B gene. This leads to impaired excretion of copper from the body. Early diagnosis is needed; therefore, the risk of irreversible organ damage is minimized. The symptoms often overlap with other disorders, making it hard to identify. An evaluation clinically, blood tests and genetic screening are commonly used for the diagnosis, subsequently treatment is provided with chelating agents such as penicillamine and trientine that contribute to removing copper. Zinc salts may be added at some times to reduce copper absorption. Liver transplantation may be done in severe liver damage. Throughout the article, the topics discussed are the cause, clinical features, diagnostic methods, therapy and future direction of management.
Wilson disease (WD) is an unusual inherited deficiency in copper metabolism. It was first described by William Alexander Kinnier Wilson, British neurologist, in 1912 [1–3]. Symptoms of WD include progressive copper accumulation predominantly in the liver, brain, cornea, and other organs; it can cause a wide variety of hepatic, neurological, and psychiatric disorders. If left untreated, the liver will become severely damaged; neuropsychiatric symptoms will become severe and persistent, affecting daily life and eventually causing death [3–6]. Because WD produces variable clinical presentations, it is often mistakenly diagnosed or diagnosed after its initial symptoms occur; earlier detection and treatment are therefore essential to prevent irreversible organ damage [1,4,7]. Epidemiologically, Wilson disease is estimated to be in the world prevalence of 1 in 30, 000 to 1 in 50, 000 individuals, although the carriers of the heterozygous mutations in the gene responsible for ATP7B are much more common than this, approximately 1 in 90 in most populations [1,3,4,6,8]. The potential underreporting of the prevalence is thought to contribute to a general underdiagnosis of WD, especially in developing countries where access to specialized diagnostic facilities is generally poor. Paternal studies have also shown that WD is not universally common; in some regions, such as Eastern Europe, India, China, and the Middle East, the prevalence rate is relatively higher (thanks in part to consanguinity and founder mutations) [3,7]. In addition, in many countries it occurs more severe in childhood. In this population many patients develop liver or cardiovascular disease as young as 5 years of age, while neurological and psychological symptoms usually appear in the teen years and early adulthood [2,4,5,9].
Yet because Wilson disease is rare, but highly inflammatory and devastating it require urgent further public awareness and treatment. The purpose of this review article is to address these issues: First, since WD is treatable, delayed diagnosis and therapy have been shown to be associated with organ dysfunction that can influence quality of life and survival. Second, the current treatment paradigm for WD has changed markedly in recent years, with the arrival of novel therapeutics such as ALXN1840 (formerly WTX101) that are expected to change how WD is treated [10,11]. Third, major advances have been made in genetic testing, biomarker discovery, and potential gene therapy, and a comprehensive review is warranted to reflect the current state of knowledge and the future direction for the treatment of this disease.
Currently, the cornerstone of WD management includes copper chelators such as penicillamine and trientine, and maintenance therapy with zinc salts [5,12,13]. However, conventional therapies are not without limitations, they are often associated with adverse effects, require lifelong adherence, and may not fully reverse neurological damage. Liver transplantation remains a last resort for those with fulminant hepatic failure or end-stage liver disease [14]. In this context, new therapeutic approaches aiming to offer improved efficacy, better tolerability, and even curative outcomes are emerging. A significant milestone in this regard is the clinical evaluation of ALXN1840, a high-affinity copper-binding molecule, which has shown promising results in clinical trials by achieving effective copper control with a once-daily dosing regimen and a favorable safety profile compared to traditional chelators [6,9,10].
A comprehensive review of Wilson disease is also timely because recent studies have broadened our understanding of its complex genetic underpinnings. Mutations in ATP7B are known to be highly heterogeneous, with more than 500 pathogenic variants identified to date [9]. This genetic diversity contributes to the clinical variability seen in patients, posing diagnostic challenges. Furthermore, the increasing accessibility and decreasing costs of genetic testing now allow for earlier and more accurate diagnosis, enabling presymptomatic treatment in at-risk individuals. Nevertheless, challenges remain in the interpretation of novel variants of uncertain significance, necessitating further research and consensus in genetic diagnostics.
Another area undergoing transformation is the field of diagnostic biomarkers. Traditionally, low serum ceruloplasmin levels and an elevated 24-hour urinary copper excretion have been the standard biomarkers of this condition [15]. However, these biomarkers can be difficult to interpret due to the poor sensitivity and specificity. A range of novel diagnostic strategies such as direct quantification of the non-ceruloplasmin binding (“free”) copper, copper isotope analysis, combined with advanced imaging techniques (quantitative MRI) have been proposed [15]. To further study future directions, gene therapy is a promising topic, with preclinical models demonstrating the pharmacological feasibility of treatment by AAV-directed gene replacement therapy (ATP7B, 1 in 11). This may provide a curative one-time therapy, as there is significant evidence that gene therapy could impact the overall course of WD in a way that does not necessitate lifelong therapy. Also, the emphasis on patient-centred research regarding quality of life and adhesion to therapy, and mental health outcomes is growing in this understanding that management for WD is in many respects broader than biochemical control.
Studies since 2010 present a growing interest in the comprehensive management of Wilson disease. For instance, in a systematic review by Schilsky et al., the need for personalized therapeutic treatments depends upon the phenotype, mutation profile, and patient preferences. A greater attention is being paid to real-world studies reporting treatment outcome and developing natural history registers for WD that facilitate evidence-based practice. However, with respect to all of this, few further steps have been taken to standardize diagnostic criteria, optimal algorithms for neurological Wilson disease treatment, or fetal management, which require further research and consensus building [1,2].
Summary as mentioned in the title, Wilson disease is a rare model of a treatable genetic disorder where prompt diagnosis and effective disease management can dramatically modify the outcome of patients. The current state of diagnostics and therapies as well as the potential use of new technologies, such as gene therapy, make a review of existing knowledge the most important step towards developing new treatment strategies for Wilson disease. In this review article we aim to provide an updated review of the causes, diagnosis, treatment options, and future directions of Wilson disease, considering the latest clinical trials, translational research, and expert guidelines to provide a comprehensive resource for clinicians, researchers, and students, as well as a source of inspiration for future research which will contribute towards further improvements in the outcomes of patients with Wilson disease.
Causes and Pathophysiology of Wilson Disease
Wilson disease (WD) arises due to pathogenic mutations in the ATP7B gene, located on chromosome 13q14.3 [1,9,16,17]. The ATP7B gene encodes a copper-transporting P-type ATPase, a transmembrane protein critical for two major physiological processes in hepatocytes: the incorporation of copper into apo-ceruloplasmin to form holo-ceruloplasmin and the excretion of excess copper into bile. When ATP7B function is compromised due to mutations, copper homeostasis is disrupted, leading to copper accumulation first in the liver, followed by its spillover into the bloodstream and deposition into other organs such as the brain, kidneys, and cornea [6,9,16–18] (Figure 1).
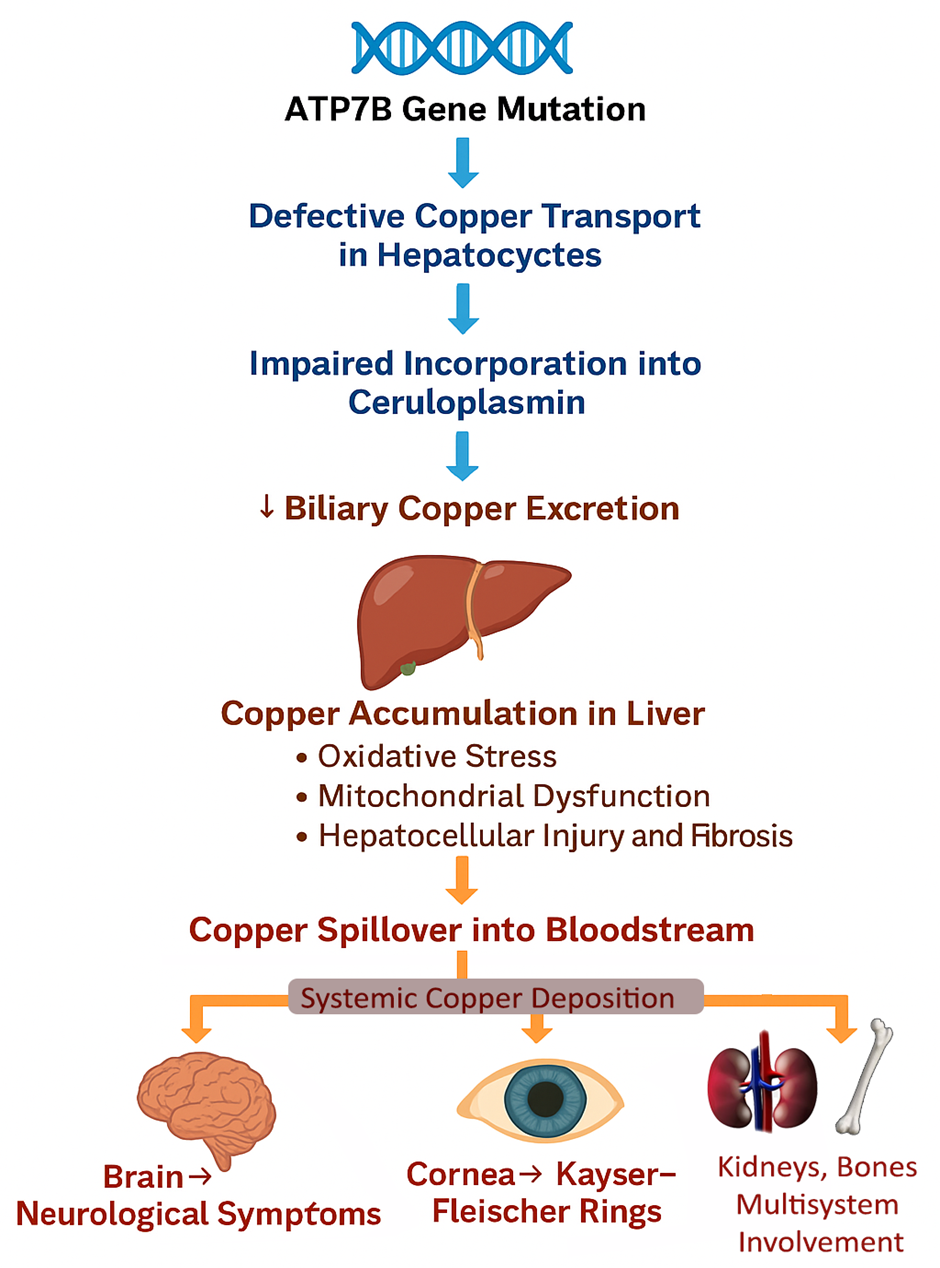
Figure1: Pathophysiology of Wilson Disease
Genetic Basis
Over 500 mutations in the ATP7B gene have been reported, most of which are missense mutations, although nonsense mutations, insertions, deletions, and splice-site mutations also occur [9]. The most common mutation in European populations is H1069Q, while other variants such as R778L predominate in East Asian populations [9,19]. The wide range of mutations correlates with phenotypic diversity, where some mutations result in primarily hepatic disease and others lead to neurological manifestations [16,17].
WD follows an autosomal recessive inheritance pattern; individuals must inherit two pathogenic ATP7B alleles (homozygous or compound heterozygous) to develop the disease. Carriers of a single mutation (heterozygotes) are typically asymptomatic but may show subtle abnormalities in copper metabolism [16,17].
Pathophysiology of Copper Accumulation
In healthy individuals, excess dietary copper is excreted through bile. In WD patients, defective ATP7B function prevents biliary excretion, resulting in toxic copper accumulation within hepatocytes [6]. Over time, copper-induced oxidative stress leads to hepatocellular injury, inflammation, fibrosis, and cirrhosis. When hepatic copper-binding capacity is overwhelmed, non-ceruloplasmin-bound ("free") copper is released into the bloodstream, contributing to systemic copper toxicity [6,9,17].
The brain, especially the basal ganglia, is highly susceptible to copper-induced neurotoxicity. Copper promotes the formation of reactive oxygen species (ROS), impairs mitochondrial function, and induces apoptosis of neurons, leading to the characteristic neurological and psychiatric manifestations of WD [16,17].
Copper also deposits in the kidneys, bones, and cornea (notably forming Kayser-Fleischer rings in Descemet's membrane), further contributing to the multi-organ impact of the disease. Key pathogenic mechanisms include oxidative stress, where the production of reactive oxygen species damages lipids, proteins, and DNA, leading to cellular injury. Mitochondrial dysfunction also plays a critical role, as it impairs ATP production and triggers apoptosis, contributing to cell death. Additionally, inflammatory pathways are activated, resulting in the stimulation of hepatic stellate cells and the promotion of fibrogenesis. Lastly, neuronal degeneration occurs due to the direct toxic effects on neurons and glial cells, further exacerbating tissue damage and dysfunction [6,16,17].
Clinical Correlation
The extent and distribution of copper deposition largely determine the clinical presentation. Hepatic symptoms often precede neurological symptoms in paediatric cases, while young adults may present primarily with movement disorders, psychiatric symptoms, or a combination thereof [1,2,8]. Interestingly, some individuals with severe ATP7B mutations may remain asymptomatic into adulthood, suggesting the existence of modifying genes, epigenetic factors, or environmental influences that modulate disease expression.
Diagnosis of Wilson Disease
Early and accurate diagnosis of Wilson disease (WD) is crucial for initiating timely treatment and preventing irreversible organ damage. However, the diagnosis remains challenging due to the heterogeneity of clinical presentations, ranging from asymptomatic liver enzyme elevation to fulminant hepatic failure, or isolated neuropsychiatric symptoms without overt liver dysfunction [2,8,9,15]. A combination of clinical evaluation, biochemical testing, ophthalmologic examination, imaging, and genetic analysis is typically required for a definitive diagnosis shown in table 1.
Table:1 Diagnosis of Wilson Disease [15,20,21]
|
Category |
Test/Feature |
Description |
Key Findings |
Limitations |
|
Clinical Suspicion |
General Indicators |
Unexplained liver, neurological, psychiatric issues in individuals <40 years |
Liver dysfunction, tremors, psychiatric changes, hemolytic anemia |
Particularly strong suspicion if both hepatic and neurological symptoms coexist |
|
Biochemical Testing |
Serum Ceruloplasmin |
Copper-carrying protein produced by the liver |
Level <20 mg/dL suggests WD |
Low in other conditions (malnutrition, protein loss); may be normal during inflammation or acute liver failure |
|
24-hour Urinary Copper Excretion |
Measures urinary copper output |
>100 µg/24 hours is diagnostic; often markedly elevated in symptomatic patients |
Repeat testing or stimulation with penicillamine may be needed in asymptomatic or early cases |
|
|
Hepatic Copper Quantification |
Measures copper in liver tissue from biopsy |
>250 µg/g dry weight confirms WD |
Sampling errors possible; cholestasis can falsely elevate copper levels |
|
|
Ophthalmologic Examination |
Slit-lamp Exam |
Detects copper deposits in the eye |
Kayser-Fleischer rings present in almost all neurological cases and 50–60% hepatic cases |
Sunflower cataracts are less common but also indicative |
|
Imaging Studies |
Brain MRI |
Assesses brain changes in neurological WD |
T2 hyperintensities (basal ganglia, thalamus, brainstem, cerebellum); "Face of the giant panda" sign; brain atrophy in late stages |
MRI findings support but are not specific for WD |
|
Abdominal Imaging |
Ultrasound, MRI, or elastography of the liver |
Signs of chronic liver disease, cirrhosis; noninvasive fibrosis assessment |
Findings are non-specific; imaging complements other tests |
|
|
Genetic Testing |
ATP7B Gene Analysis |
Identifies mutations causing WD |
Confirms diagnosis; allows family screening |
Variants of uncertain significance (VUS) require clinical and biochemical correlation |
Diagnostic Scoring Systems
The Leipzig (Ferenci) scoring system is widely used, assigning points to clinical, biochemical, ophthalmological, histological, and genetic findings [21] (Table 2).
Table 2: Leipzig (Ferenci) scoring system [15,22]
|
Parameter |
Score |
|
Kayser-Fleischer ring present |
2 |
|
Neurological symptoms present |
2 |
|
Serum ceruloplasmin <0.2 g/L |
1 |
|
24-hour urinary copper >1.6 µmol |
1 |
|
Liver copper >4 µmol/g dry weight |
2 |
|
Presence of ATP7B mutations (2 mutations) |
4 |
|
Presence of ATP7B mutations (1 mutation) |
1 |
Diagnostic Challenges: Despite the availability of multiple diagnostic tools, Wilson disease remains underdiagnosed or misdiagnosed due to several factors. The clinical presentation of the disease is highly variable, often mimicking other liver, psychiatric, or neurological disorders, which can lead to confusion and delayed diagnosis. Additionally, individual diagnostic tests have their own limitations, making it difficult to rely on a single test for confirmation. A further challenge is the lack of awareness about Wilson disease among general physicians and non-specialists, which contributes significantly to missed or incorrect diagnoses. Timely diagnosis depends on maintaining a high index of suspicion and employing a multimodal diagnostic approach.
Treatment Options for Wilson Disease
The primary goal in the treatment of Wilson disease (WD) is to remove excess copper from the body, prevent its reaccumulation, and reverse or stabilize the manifestations of the disease. Early diagnosis and lifelong adherence to treatment are essential to achieve favorable outcomes[6,11,18]. Therapeutic strategies include copper chelation, zinc therapy to block copper absorption, liver transplantation in advanced disease, and, more recently, novel agents like ALXN1840 and summary shown in table 3.
1. Chelating Agents
Chelators promote the excretion of stored copper through the urine. Two main chelators are currently approved.
Penicillamine: Penicillamine was the first effective oral treatment introduced for Wilson disease. It works by binding to copper and enhancing its urinary excretion. Treatment typically begins with a low dose, such as 250 mg/day, and is gradually increased to a maintenance dose of 750–1500 mg/day to minimize the risk of adverse effects [12,13,20,23]. Regular monitoring is essential, including assessments of urinary copper levels, complete blood counts, and renal function. Despite its effectiveness, penicillamine is associated with significant adverse effects, including hypersensitivity reactions, nephrotoxicity, bone marrow suppression, and paradoxical neurological worsening, particularly if high doses are initiated too quickly. Due to these serious side effects, many patients are unable to tolerate penicillamine, leading to the need for alternative therapies [12,13,20,23].
Trientine: Trientine is a newer chelating agent used as a first-line or alternative therapy for Wilson disease, particularly in patients who are intolerant to penicillamine. It is typically administered at doses ranging from 750 to 1500 mg/day, divided throughout the day [13,23]. Trientine offers advantages over penicillamine, including a lower incidence of hypersensitivity reactions and overall better tolerability. However, it is not without adverse effects, which can include iron deficiency anemia, gastrointestinal upset, and, in rare cases, neurological worsening. A special consideration with trientine is that it must be taken on an empty stomach to maximize absorption. Importantly, both penicillamine and trientine require close clinical and biochemical monitoring to avoid complications related to overtreatment, such as copper deficiency, or undertreatment, leading to persistent copper overload [13,23].
2. Zinc Therapy
Zinc acts by inducing the production of metallothionein in enterocytes, which binds dietary copper and prevents its absorption into the bloodstream; the copper-laden enterocytes are then naturally shed into the intestinal lumen and eliminated in feces [12,13,20]. Zinc is indicated for maintenance therapy following initial chelation and as a first-line treatment for presymptomatic or asymptomatic patients. The recommended dosing is 150 mg of elemental zinc daily, divided into three doses. While generally well-tolerated, zinc can cause adverse effects such as gastric irritation, nausea, and, rarely, pancreatic dysfunction. Regular monitoring of urinary copper excretion and serum copper levels is crucial to ensure therapeutic effectiveness and avoid complications. Zinc therapy is particularly attractive due to its relative safety, ease of oral administration, and effectiveness in maintaining long-term copper balance, especially in asymptomatic individuals and during pregnancy [12,13,20].
3. Liver Transplantation
Liver transplantation is a life-saving intervention for patients with Wilson disease who develop acute liver failure, often presenting as fulminant hepatic failure, or for those with end-stage liver disease unresponsive to medical therapy. The procedure corrects the underlying defect by replacing the defective ATP7B protein with a functioning one from the donor liver, thereby restoring normal copper metabolism [9,14]. Survival rates following transplantation are excellent, with one-year survival exceeding 85% in most centers. However, in patients with predominantly neurological symptoms but compensated liver function, liver transplantation is generally not effective for neurological recovery and is reserved primarily for cases with significant hepatic involvement [14].
4. Emerging Therapies: ALXN1840
ALXN1840 (formerly known as bis-choline tetrathiomolybdate) represents a promising advancement in the treatment of Wilson disease. It is a novel copper-binding agent that works by forming a stable tripartite complex with protein-bound copper and albumin, facilitating copper removal while minimizing the toxicity caused by free copper. Clinical trials, particularly the FOCUS study, have shown that ALXN1840 provides superior control of non-ceruloplasmin-bound copper compared to standard treatments like penicillamine or trientine after 48 weeks of therapy. Notably, ALXN1840 demonstrated particular effectiveness in patients with neurological manifestations, achieving improvements without the paradoxical neurological worsening often seen with traditional chelators. Its advantages include once-daily oral dosing, a favorable safety profile, and a reduced risk of neurological deterioration. ALXN1840 is currently undergoing regulatory evaluation, with potential approval expected in many regions. If approved, it could revolutionize Wilson disease management by offering a safer, more effective, and more convenient therapeutic option [10,11].
Table:3 Summary of Treatment Options for Wilson Disease [13,20]
|
Treatment Option |
Mechanism of Action |
Indication |
Notes/Side Effects |
|
Penicillamine |
Chelates copper to promote urinary excretion |
First-line for symptomatic patients |
Risk of hypersensitivity, nephrotoxicity, bone marrow suppression |
|
Trientine |
Chelates copper to enhance urinary excretion |
Alternative for patients intolerant to penicillamine |
Fewer side effects than penicillamine but may cause anemia |
|
Zinc salts (e.g., zinc acetate) |
Blocks intestinal copper absorption |
Maintenance therapy; presymptomatic or mild disease |
Can cause gastrointestinal irritation; requires lifelong use |
|
Liver Transplantation [14] |
Replaces damaged liver, restores copper metabolism |
Fulminant hepatic failure or end-stage liver disease |
Lifesaving but involves lifelong immunosuppression |
|
Dietary Modification |
Reduces copper intake (avoiding high-copper foods) |
Adjunct to medical therapy |
Not sufficient alone; includes avoiding shellfish, nuts, chocolate, mushrooms |
|
Experimental Therapies (Gene Therapy, Novel Chelators) |
Corrects genetic defect or improves copper binding |
Future potential treatments |
Still under investigation in clinical trials |
5. Dietary Copper Restriction
Although pharmacological therapy remains the cornerstone of Wilson disease management, dietary modifications are also recommended, especially during the early stages of treatment. Patients are advised to avoid copper-rich foods such as shellfish, nuts, chocolate, mushrooms, organ meats like liver, and drinking water from copper pipes. Strict copper restriction is particularly important during the initial phase of therapy to help reduce copper overload, but dietary guidelines can often be relaxed once copper levels are adequately controlled through medical treatment.
Future Directions in the Management of Wilson Disease
While advances in diagnostics and therapeutics have dramatically improved outcomes for patients with Wilson disease (WD), several unmet needs remain, particularly regarding early diagnosis, individualized therapy, and long-term disease management. Emerging research offers promising new avenues for improving patient care and possibly achieving a cure. Future strategies in the Management of Wilson Disease shown in figure 2.
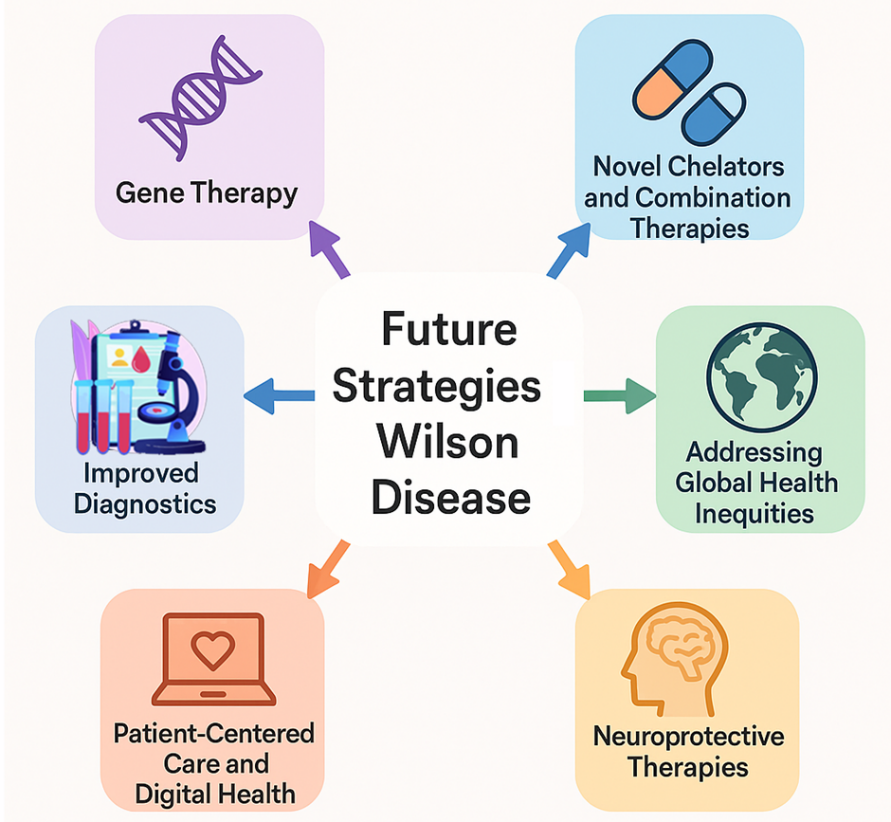
Figure 2: Future strategies in the Management of Wilson Disease
1. Gene Therapy: Since Wilson disease is caused by mutations in the ATP7B gene, gene therapy offers a logical and potentially curative approach. Preclinical studies have shown promising results, with adeno-associated virus (AAV) vectors successfully delivering functional copies of the ATP7B gene to hepatocytes in animal models, thereby restoring copper excretion and reducing liver damage [9]. Unlike lifelong chelation or zinc therapy, gene therapy has the potential to correct the underlying genetic defect with a single administration. However, several challenges remain, including the need to establish long-term safety and efficacy, address concerns about vector immunogenicity and optimal dosing, and ensure global access and affordability once approved. Several clinical trials focused on ATP7B gene delivery are expected to begin in the coming years, representing a potential turning point in the future management of Wilson disease.
2. New Chelators and Combination Therapies: While chelators for copper are effective, they are often inconvenient and tainted by toxicity and patient compliance. In the future, newer (second-generation) chelators might have greater copper specificity, fewer side effects and more convenient dosing instructions [11,18]. Combination therapies, such as the use of a chelator with zinc, may allow for even better treatment by increasing removal of copper and inactivating its absorption. The ALXN1840 antibody family and other agents have the potential to create a new class of agents to maintain copper levels as well as minimize neurologic damage [10,11]. These findings could revolutionize how Wilson disease is treated. They might lead to personalized, safer and more effective treatment strategies, providing long-term therapy options for Wilson disease patients.
3. Improved Diagnostic Techniques: While there are a number of well-established biochemical and clinical markers for Wilson disease, the condition remains undiagnosed until significant (sometimes irreversible) damage has occurred. In future research, the need for novel therapeutic agents to detect preclinical copper dysregulation may be addressed by developing highly sensitive biomarkers that can detect copper dysregulation early (before clinical symptoms take hold). In addition, newborn screening using dried blood spot testing to detect mutations in ATP7B or other copper metabolism biomarkers may be expanded. An alternative approach is the use of imaging, such as quantitative susceptibility mapping (QSM) MRI, that uses noninvasive techniques to localize accumulated copper in the brain at relatively low levels of risk.
4. Pharmacogenomics and Individualized Therapy: It is clear that individualized therapy targeting the individual’s specific mutations and copper metabolism as well as the treatment responses of the disease in question, by way of genome-phenotype correlation studies, could provide additional information about the severity of Wilson disease and determine more appropriate treatments. Genotype-phenotype correlation studies continue, with further targets to guide the assessment of disease severity and prognosis through clinical outcomes as well as to provide insight into the potential for pharmacogenomic profiling to help clinicians select the appropriate initial therapy (such as a choice between a chelator or zinc) for each individual with Wilson disease.
5. Addressing Global Health Inequities: Despite its rarity, Wilson disease continues to be a highly unequal disease globally. In future work, efforts should be focused on raising awareness of this disorder among health care professionals in developing countries to ensure these professionals are adequately trained to detect this disease early in the disease spectrum. Development of cost-effective diagnostic tools suitable for use in resource-limited settings is also key for early detection in this disease, and further reduction in barriers to treatment in disadvantaged countries, for example through generic availability of chelators and zinc salts. International collaborations and public health initiatives will be essential in bridging these gaps, ensuring that advances in Wilson disease management reach patients globally, not just those in high-income countries.
6. Neuroprotective Therapies: Chelation therapy in patients with neurological manifestations of Wilson disease is ineffective when copper levels in the brain are high. Future research will focus on several promising approaches to maximizing the outcome for patients with severe neuropsychiatric features[1,8]. Neuroprotective agents are being investigated in patients with relatively advanced neurological signs. These include antioxidants and anti-inflammatory drugs. Brain-targeted chelators have also been developed and are currently being studied in patients with persistent neurological symptoms to maximize the effectiveness and safety of treatment. Brain -targeted chelators may also be beneficial to treat central nervous system copper accumulation in patients with mild or severe neurological symptoms. Rehabilitative interventions are being considered in patients with advanced neuropsychiatric symptoms, such as physical therapy, occupational therapy, and cognitive rehabilitation [24]. These treatment strategies may also significantly improve outcome and quality of life in people with neuropsychiatric involvement.
7. Patient-Centered Care and Digital Health: The use of telehealth and remote monitoring will be one potential delivery mechanism for improving the patient experience and optimizing outcomes in the future. Router monitoring products and services are widely available and include, but are not limited to, mobile apps, web portals and online information resources for monitoring medication adherence and clinical symptoms, telephone consultations and visits to specialist clinics. Telemedicine consultation is another avenue to access expert and specialised care, especially for patients living in rural or remote locations, and providing expert guidance wherever they may be. The use of patient data and real-world evidence will allow insight into long-term outcomes of treatment delivery, thereby improving treatment management recommendations as well as support for patients.
CONCLUSION:
Wilson disease is still a difficult and challenging condition to treat, as it can have variable clinical presentations and some patients may develop severe organ dysfunction, which might require long-term treatment. The development of genetic screening for the disorder and early diagnosis of the condition has resulted in the provision of early therapy, which improves their prognosis in long term terms. There are still many existing treatments that can be used, including copper chelation, liver transplantation, and other therapies for copper deficiency. However, more new therapeutic approaches, such as gene therapy and targeted options, exist to tackle more effectively the systemic mechanisms of copper accumulation. This research is going on, and this field is one which may help us advance the biology of this disease.
REFERENCES

Sandeep Bolla, Wilson Disease: Causes, Diagnosis, Treatment Options, and Future Directions, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 6, 3387-3399. https://doi.org/10.5281/zenodo.15718234
 10.5281/zenodo.15718234
10.5281/zenodo.15718234
