Director, Statistical Programming and Research & Development - Statistics and Data Management Raleigh, United States
The importance of drug development and discovery in the biopharmaceutical industry is growing. Nowadays, the process of developing new drugs is laborious, expensive, ineffective, and prone to errors. A very valuable method for drug discovery applications might be a digital microfluidic platform. Algorithms for ML have also shown to be a helpful tool for drug development. This study develops and evaluates various ML models, specifically SVM and ANN, to classify compound activity effectively. These models were created and assessed using the ChEMBL dataset, and performance metrics such as accuracy, precision, recall, and F1-score were used. The findings were obtained using confusion matrices. The ANN model demonstrated a remarkable accuracy of 91%, complemented by high precision 95% and recall of 87%, indicating robust predictive capabilities. In comparison, the SVM model exhibited a lower accuracy of 70%, highlighting the ANN's superior ability to capture complex patterns. These outcomes underscore the significant potential of ML, particularly ANN, in optimizing drug discovery processes and facilitating more effective identification of active compounds.
The foundation of the pharmaceutical sector is innovation. Innovation may provide life-saving solutions and give society access to life-saving medications. Thus, developing and patenting novel medicinal molecules is crucial. In today's scientific world, research plays a vital role in the discovery, testing, and eventual patenting of new kinds of pharmaceuticals [1][2][3]. This approach is being used by the pharmaceutical industry to develop and patent novel medications. New medications for diseases, including neglected diseases, are found via the drug development process. A significant amount of money is being invested in this direction by pharmaceutical corporations. Nonetheless, the process of finding and developing new drugs is quite intricate. Drug research and discovery have made use of a wide range of technology and a high degree of competence [4]. The time required for drug discovery is significant. As a result, the drug discovery industry has seen the emergence of a new, creative technical environment. Faster drug discovery techniques are one way that innovative technologies benefit the pharmaceutical industry. Drug research and discovery may be greatly impacted by recent developments in cutting-edge instruments and technology.
Today's breakthrough techniques and technology have accelerated medication research and development. As a result, new technologies enable speedier medication discovery. Simultaneously, the technique increases productivity in the pharmaceutical business. This is crucial for the pharmaceutical industry. Meanwhile, pharmaceutical companies need to step up their game when it comes to developing new drugs. A highly efficient drug discovery method may be rapidly developed with the use of innovative technology[5][6][7].
Research and development of new drugs entails three primary phases: identification of target molecules, creation of drug molecules for use in preclinical studies, and finally, the advancement of these molecules to a clinical trial. There are a number of problems with the traditional approaches to drug discovery and development. They are inefficient, take a lot of time, and don't work very well. An estimated US$2.6 billion and ten to fifteen years are required to bring a brand-new medicinal compound to market [8][9][10]. The capital cost of developing new drugs is US$1.3, according to a recent cost study. Having said that, the average amount needed to achieve success is only $200 million. Also, $1 billion USD is spent on failures that are paid for out of pocket. Consequently, lowering the failure costs may lower the cost of discovering a successful medication. New technologies like AI may lower the failure costs of drug research[11].
In the last ten years, artificial intelligence has been used in several fields of the general, biological, and medical sciences. AI, sometimes known as machine intelligence, governs how computers may learn from past data and user interaction [12]. Analyses related to drug discovery and development are connected with several AI techniques[13][14], including conventional ML[15] and DL[16]. Contrarily, a substantial step in the process of generating new drugs was creating various models. Hence, model architectures have also been developed to lower healthcare costs.
Motivation and Contribution of paper
The motivation behind this study lies in addressing the growing complexity and time-intensive nature of pharmaceutical drug discovery. Traditional drug discovery methods can be costly, slow, and often have low success rates due to the challenges in accurately predicting compound bioactivity and potential therapeutic efficacy. This study contributes to the field by developing a machine learning-based framework that enhances bioactivity prediction and optimizes pharmaceutical drug discovery. The main contributions are:
Utilized the ChEMBL database, containing over 1.9 million compounds.
Implemented comprehensive data preprocessing steps, including removal of missing and duplicate entries, to ensure high-quality input for the model.
The model's capacity to generalize across classes of bioactivities was improved, leading to better overall performance by resolving class imbalance using SMOTE.
Metrics for evaluating models' performance, like F1-score, recall, accuracy, and precision.
Offered a scalable and adaptable approach that can be applied to diverse datasets in drug discovery.
Organization of paper
Here is how the rest of the paper is organized: Background information on drug discovery is given in section II. Section III provides the research methods used in this work. Section IV provides a comprehensive overview of the results, model descriptions, and commentary. Section V concludes by discussing the study's contents, limitations, and potential future research paths.
Related Work
This section presents previous research on pharmaceutical drug discovery using machine-learning approaches. Some of the research are:
This research paper, Aluvala et al., (2024) uses a large drug network to create a ML model that forecasts a drug's effect. The model integrates the J-48 algorithm and random forest algorithm and forms the hybrid bagging technique with the neural network model demonstration. The study’s findings highlight the importance of machine learning models in drug classification, which offers a valuable tool for researchers and pharmaceutical companies in the drug development process. The output includes the top classes of drugs, the prediction of action and aiding in new drug discovery with a higher accuracy of 95.83%, which is superior to other learning models[17].
In, Choudhari et al., (2024) utilised Random Forest nearby other calculations to foresee potential medicate targets. Our comprehensive assessment measurements, counting F1 score, recall, accuracy, and precision, emphasize the prevalent execution of Random Forest in comparison to DT, SVM, and KNN. The outcomes demonstrate a precision of 85%, accuracy of 86%, review of 84%, and an F1 score of 85% for Irregular Forest, certifying its viability in precisely recognizing promising sedate targets. The study contributes to the advancing scene of machine learning applications in biomedical research, emphasizing the potential of gathering learning procedures, especially Irregular Woodland, to streamline and improve the target identification stage of sedate improvement[18].
This paper, Gawich and Alfonse, (2022) offers a model that uses ML algorithms and NLP methods to forecast medication ratings based on review analysis. By leveraging the R-program's Syuzhet package to extract sentiment from reviews, the model facilitates the finding of medications that cause adverse drug reactions. The suggested model passes the Drugs.com and DrugsLib.com tests with an accuracy of 87% and 78%, respectively. Additionally, the model was used to find the ADRs on the Drugs.com dataset. Additionally, 66 reviews out of 213865 show that people experience serious adverse effects. ADRs are thought to be triggered by both mild and severe adverse effects. Put another way, 21.03% of medication evaluations state that the patient had adverse pharmacological reactions as a result of using the medication[19].
In, Zamitalo et al., (2022) developed a method to anticipate COVID-19 drug targets using ML to quickly identify possible drugs for 18 COVID-19 protein targets. We focused on three prediction models' abilities to foretell drug-target docking scores, which are a measure of the strength of interactions between ligands and proteins, with figures like R2=0.69, MAE=0.285, and MSE=0.627, our proposed technique performed competitively. We also discover chemical combinations that are associated with greater binding affinities at target binding sites. They think that our research may help pharmaceutical firms save a lot of money, particularly in the early phases of medication development[20].
In this paper, Hu et al., (2021) suggested a brand-new DL-based prediction technique to find drug-target interactions (DTIs) with a new negative instance generation. Consequently, our approach produced a dataset with an accuracy of 0.9800. The model's generalization was further evaluated using a different dataset that was taken from Drug Bank. The model performed well on the dataset, with an accuracy of 0.8814 and an AUC value of 0.9527. Our experimental findings showed that the suggested approach, which uses believable negative creation, may be used to distinguish between drug-target interactions. Website: http://www.dlearningapp.com/web/DrugCNN.htm[21].
This paper, Saber, Rihana and Mhanna, (2019) shows two BBB models—an in vitro model and an in silico model—that were created and used for early-stage drug research. The previous model achieved a maximum overall accuracy of 96.23% using SVM and QDA classifiers after a genetic method was used for feature selection. To improve the endothelial cells' adherence to the substrate, several coatings were explored. The collagen type I covering produced the greatest level of confluency. Additionally, the highest trans-endothelial electric resistance (TEER) value measured was 45.6 ± 12.07 ?.cm 2, which is comparable to readings reported using the same cell line[22].
This study, Bahi and Batouche, (2018) DL algorithms may be used to search through huge databases of chemicals to classify ligands as either drug-like or non-drug-like against a certain protein target, which helps speed up the VS process. In order to classify tiny compounds from large datasets, this research suggests a quick compound classification technique dubbed DNN-VS, which is built on a DNN for Virtual Screening and uses the Spark-H2O platform. According to experimental data, the suggested method works better than the most advanced ML approaches, with an overall accuracy of over 99%[23].
Table I summarizes the information from each research paper based on authors, data, approach, findings, and limitations/contributions.
Table 1 Summary of Machine Learning Approaches in Drug Discovery, Development, and Target Identification
|
Authors |
Data |
Approach |
Findings |
Limitation/Contribution |
|
Aluvala et al., 2024 |
Large drug network, Struck2Vec |
Hybrid bagging technique (J-48, Random Forest, Neural Network) |
Achieved 95.83?curacy in drug classification, aiding drug discovery. |
Demonstrated a highly accurate model for drug classification, enhancing drug development. |
|
Choudhari et al., 2024 |
Drug datasets |
Random Forest, Decision Trees, SVM, k-NN |
RF achieved accuracy: 86%, precision: 85%, recall: 84%, F1: 85%, effective for drug target identification. |
Validates the potential of RF for drug target identification, aiding in drug discovery processes. |
|
Gawich and Alfonse, 2022 |
Drugs.com dataset, DrugsLib |
Sentiment analysis, NLP (syuzhet in R) |
87?curacy (Drugs.com), 78% (DrugsLib); identified ADRs from patient reviews. |
Effective in detecting ADRs from reviews, offering insights for patient safety and drug monitoring. |
|
Zamitalo et al., 2022 |
COVID-19 protein docking targets |
ML models for docking score prediction |
R?2;: 0.69, MAE: 0.285, MSE: 0.627; identified strong binding affinities for COVID-19 targets. |
Offers cost-effective support for early drug discovery in pandemic scenarios. |
|
Hu et al., 2021 |
DrugBank-derived dataset |
Deep learning with negative instance generation for DTI prediction |
Accuracy: 0.9800 (created dataset), 0.8814 (DrugBank); AUC: 0.9527, effective DTI discrimination. |
Effective DTI identification, contributing to drug-target interaction research with high accuracy. |
|
Saber, Rihana, and Mhanna, 2019 |
In silico and in vitro BBB models |
Genetic Algorithm, QDA, SVM, PLA 3D inserts |
96.23?curacy; tested coatings for cell adhesion, achieved high TEER for in vitro BBB model. |
Low-cost BBB model validation for drug screening, supporting cost-effective drug testing methods. |
|
Bahi and Batouche, 2018 |
Large molecular databases |
Deep neural network for virtual screening (DNN-VS, Spark-H2O) |
Over 99?curacy in ligand classification for VS, efficient database screening. |
Enhances VS by rapid classification, valuable for pharmaceutical research and large-scale molecular screening. |
Methodology
The methodology for applying machine learning to optimize pharmaceutical drug discovery involves several key steps. The following workflow of this research is shown in Figure 1. Data is first collected from sources like ChEMBL, an open-source database containing extensive bioactivity information on small molecules, including properties and biological targets. Following data collection, preprocessing steps ensure data quality, such as removing missing or duplicate values, labelling compounds based on bioactivity, and balancing classes using techniques like SMOTE to handle imbalanced data. Next, data is split into training and testing sets to validate model performance. The SVM and ANN are then applied. A confusion matrix is used to quantify the model's accuracy, precision, recall, and F1-score, which are measures used to evaluate the model's performance in classification. This process helps determine the best model parameters and evaluation metrics, ultimately aiding in the development of an efficient drug discovery model that can predict the bioactivity of new compounds with higher accuracy.
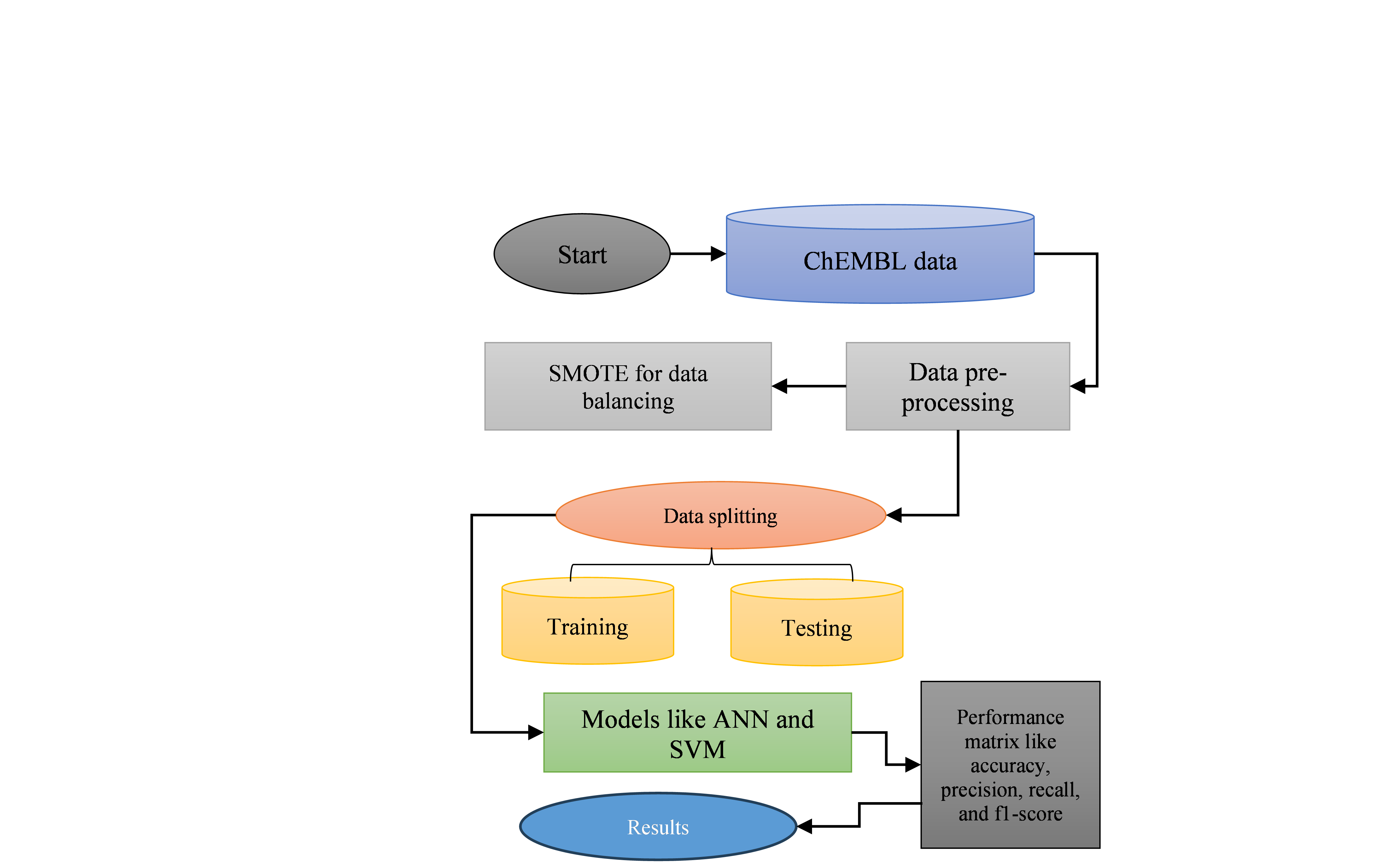
Figure 1 Flowchart for pharmaceutical drug discovery
Each step of the flowchart in Figure 1 for pharmaceutical drug discovery is briefly explained below:
Data collection
ChEMBL is a free and open-source database that contains information on the chemicals' biological characteristics. The most current update was in 2018, and since then, the database has continued to grow, presently including over 1.9 million chemical compounds. To the best of ChEMBL's knowledge, each of these substances has over 10,000 medications and over 12,000 targets. Further analysis of data is visualised below:
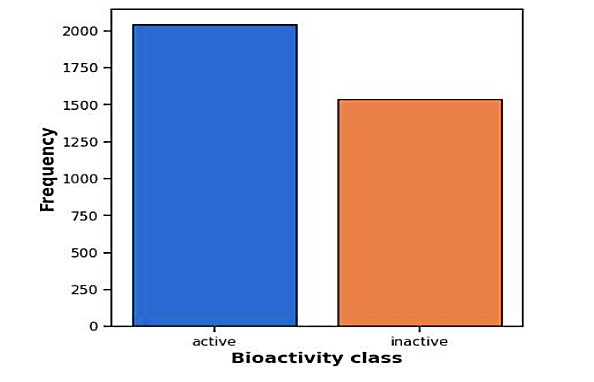
Figure 2 Frequency plot of two bioactivity classes
Figure 2 shows a frequency map of two bioactivity classes, which show how the chemicals in the dataset are distributed. It shows that there are roughly 1,500 inert compounds and around 2,000 active ones. This visual representation highlights an imbalance, with a greater number of active compounds compared to inactive ones. Such a disparity may require attention during model training to ensure that the model effectively learns from both classes.

Figure 3 Bar graph for top bioactivity units
Figure 3 shows the frequency of bioactivity units in a dataset. "Potency" is the most common unit, with around 60,000 occurrences, followed by "IC50" (approximately.
2,000), "Ki" (around 1,000), and "Kd" (about 500). This graph visually illustrates that "Potency" dominates the dataset, while the other units are significantly less frequent.
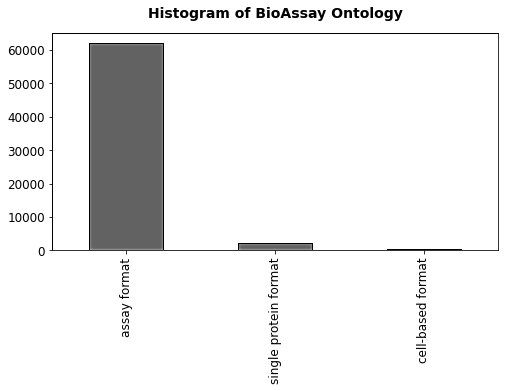
Figure 4 Histogram for bioassay ontology
The Histogram of BioAssay Ontology in Figure 4 displays the distribution of bioassays by format, indicating that the "assay format" is the most prevalent, with around 60,000 occurrences. In contrast, the "single protein format" has approximately 2,000 instances, and the "cell-based format" is represented by about 1,000. This visualization highlights the dominance of the "assay format" in the dataset, while the other two formats are significantly less common.

Figure 5 Histogram of chEMBl value
Figure 5 Histogram of ChEMBL values reveals the distribution of ChEMBL values in the dataset, characterized by a right-skewed shape with a long tail towards higher values, indicating a greater number of compounds with lower ChEMBL values. The values range from approximately 2 to 10, with the majority concentrated between 4 and 8.
Data Preprocessing
A crucial step in getting datasets ready for use in ML models is data preparation. A number of preprocessing procedures were used to clean and prepare the dataset for drug development. First, compounds that had missing values in the canonical smiles and standard value columns were eliminated from the dataset. Additionally, the dataset was cleansed of any duplicate canonical grins. The IC50 values were used to categorize the compounds as active, inactive, or intermediate. A QSAR model was trained to predict the bioactivity of novel compounds using the dataset, which had 4671 rows and 4 columns once all preprocessing processes were finished.
SMOTE for Data balancing
SMOTE has remarkable performance on small datasets. SMOTE's efficiency drastically decreases with increasing dataset size, however, since it takes longer to generate fictitious data points. Additionally, the minority class's data points are likely to overlap when false data points are created in SMOTE. The following is Smote's Equation (1):
xnew, attr=xi, attr+rand0,1x (xij, attr-xi, attr
(1)
Data Splitting
Data splitting is a process of separating a dataset into two or more datasets. First, the data is divided in an 80:20 ratio between the train and test sets.
Artificial neural network (ANN) model
An ANN consists of three layers: the input layer, the hidden layer, and the output layer[24]. A three-layer neural network looks like this: the input layer takes in information, the hidden layer analyses it and adjusts the network's weights to make it better, and the output layer presents the network's outcome as classes. The learning rule and the propagation function determine the neural network's result. A propagation function, which is represented by Eq 2, is used to influence the inputs to the jth neurone from the output of the previous neurones.
Pjt=0it×wij+b
(2)
where the propagation function is denoted by ???????? (????), the output of the previous neurone by ????????(????), the weight by ????????????, and the bias by ????. For a particular input set, the learning rule adjusts NN parameters such that the network generates a favorable outcome. The learning process adjusts the network's weights in accordance with the learning rule to improve output computation[25][26][27][28]. Several neurones are used in the ANN, and their weights are changed to increase learning rate by back-propagation of mistakes and prolonged training [29][30].
Model evaluation
The classifier's performance is determined using a variety of assessment measures [31][32]. Classification accuracy was expressed quantitatively using a confusion matrix. The confusion matrix has two dimensions: the current classes and the predicted classes. A real-life example of the class is shown in each row, while the predicted class's condition is shown in each column. In the confusion matrix, FP represents false positives, FN represents false negatives, TP represents true positives, and TN represents true negatives. Below is an explanation of the study's assessment measures.
Accuracy: The accuracy of a classifier is defined as the percentage of labels that it properly predicts relative to the total classifier labels. We may define the accuracy by (3):
Accuracy=TN + TPTP + TN + FP + FN
(3)
Precision: Precision is a statistic that evaluates how often, out of all the predictions for a given class, samples are properly categorized. The equation for it is provided by (4):
Precision=TPTP+FP
(4)
Recall: The rate at which samples are properly categorized for a certain class type, given all instances of that class type, is known as recall. The formula (5) is used to compute it.
F1=2*precision*recallprecision+recall
(5)
F1 score: The F1 score, which is a harmonic mean of the recall and precision scores, is sometimes called the F measure. Regardless matter how big a number precision or recall is, the F measure will always be closer to the smaller one. Here is a definition of the F1 score (6):
F1=2*precision*recallprecision+recall
(6)
Are following evaluation parameters evaluate the model efficiency and determine the best model accuracy for drug discovery.
Results Analysis And Discussion
The results of models for drug discovery are provided in this section. the following ML models are trained on the ChEMBL data and evaluated with a performance matrix including accuracy, f1-score, recall, and precision. The following Table II provides an ANN model performance across the performance matrix.
Performance of neural network on ChEMBL data
|
Performance matrix |
Artificial Neural network |
|
Accuracy |
91 |
|
Precision |
95 |
|
Recall |
87 |
|
F1-score |
91 |

Bar Graph for artificial neural network performance
The above Table II and Figure 6 show the ANN performance on dataset. The performance of an ANN indicates a strong predictive capability, achieving an accuracy of 91%. Precision is particularly high at 95%, suggesting that the model is effective at correctly identifying positive instances with minimal false positives. Recall, at 87%, reflects a solid ability to capture true positives, although there is some room for improvement in identifying all relevant instances. The F1-score, calculated at 91, balances precision and recall, demonstrating that the model performs well overall in maintaining a good trade-off between the two metrics.

Accuracy graph for artificial Artificial neural network
Figure 7 shows an artificial neural network achieving high accuracy (99%) over 20 epochs, with training accuracy plateauing around the 15th epoch and testing accuracy slightly lower, suggesting potential overfitting. The model performs well but may benefit from techniques like regularization to improve generalization.

Loss graph for Artificial neural network
The loss graph over 20 epochs shows that the neural network’s training and testing loss initially decreased, indicating effective learning. Training loss continues to decline, but testing loss starts to level off and even rise slightly after about 5 epochs, suggesting overfitting. To address this, techniques like early stopping, L1/L2 regularization, dropout, data augmentation, and hyperparameter tuning could improve generalization. More details about the dataset, task, model architecture, and training parameters would provide further context to refine these strategies.
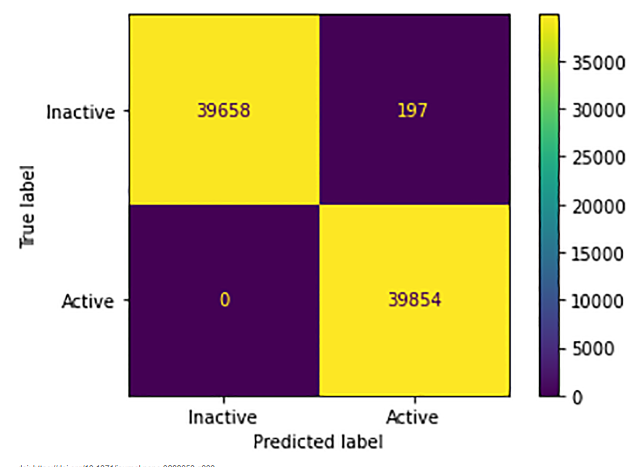
Confusion matrix for neural network
Figure 9 shows excellent model performance, with an accuracy of about 99.98%. It accurately identifies 39,658 "Inactive" and 39,854 "Active" instances, with only 197 "Inactive" misclassified as "Active" and no "Active" misclassified as "Inactive." The high precision (99.5%) and perfect recall (100%) for "Inactive" yield an F1-score of 99.75%, indicating robust classification with minimal errors. Further tuning could address the small misclassification rate, especially with additional context on dataset specifics and model parameters.
Comparison between NN and existing model performance on chEMBl data
|
Models |
Accuracy |
|
Support vector machine [33] |
70 |
|
Artificial neural network |
91 |
The comparison between models shows that the ANN significantly outperforms the SVM in terms of accuracy, achieving 91% compared to the SVM’s 70%. This suggests that the NN model is better suited for capturing the complexities of the data, while the SVM may be less effective in distinguishing between classes. This notable accuracy difference indicates that the NN might have a more appropriate architecture or greater flexibility for this specific task, making it a preferable choice in this case.
Conclusion And Future Work
The discipline of drug discovery evolved globally as a result of the introduction of bioinformatics. Drug discovery is an expensive and difficult procedure that has a poor success rate. Among the many domains that have made use of computational approaches based on ML is drug development. By leveraging the ChEMBL dataset, we developed and evaluated various ML models, specifically SVM and ANN, for predicting the bioactivity of chemical compounds. The results indicate that the ANN model significantly outperformed the SVM, achieving an accuracy of 91%, with impressive precision and recall metrics. This highlights the ANN's ability to accurately detect intricate patterns in the data, which makes it a useful tool for improving the discovery of active chemicals. Data preparation and class balancing methods, such as SMOTE, are crucial to improving model performance, as shown by the results. While the ANN showed strong predictive capabilities, further refinement through regularization and hyperparameter tuning is needed to address overfitting. The promise of ML to speed up the identification of viable pharmaceutical candidates is supported by this study, which adds to the increasing body of data supporting its use in drug development.
REFERENCES

Anilkumar Jangili, The Application of Machine Learning in Optimizing Pharmaceutical Drug Discovery, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 1696-1607. https://doi.org/10.5281/zenodo.14415417
 10.5281/zenodo.14415417
10.5281/zenodo.14415417
