Khwaja Moinuddin Chishti Language University, Faculty of Pharmacy, Lucknow, Uttar Pradesh.
Novel, highly effective, and safe treatment candidates against the wide range of disorders, such as cancer, viral infections, and microbial infections, may be discovered by structural hybridisation of pharmacologically active compounds that have already shown promise in animal studies. In an endeavour to resolve the constraints of several therapeutic treatments, strategies for drug-drug combinations have been devised to produce hybrid conjugates of various medications. Increased bioavailability, decreased toxicity, and prevention of drug resistances were the goals of developing hybrids, which are believed to enhance efficacy through likely multi-target interactions and targeted drug delivery to the site of action. In this review article, we focus on the logic of the hybrid design and the linker architecture in particular, highlighting the latest advances in the fast-expanding area of drug development. The linker architecture is critical to the overall success of a hybrid medication.
Natural products are drug-like molecules intended to be high-affinity ligands for complementary targets in biological systems, hence nature has always been driving force for drug development [1]. Developing drugs from biological and synthetic substances depends on the creation of hybrids as bi-/trivalent ligands usually show great therapeutic promise for a range of diseases [2]. In the realm of drug development, the design of these ligands has attracted much interest throughout the last two decades. Bivalent ligands have more affinity for androgenic, oestrogenic, muscarinic, and opioid receptors than their monovalent equivalents, hence generating strong interactions. Synthesised several bivalent hybrid drugs have been evaluated for their effectiveness in treating cancer, Alzheimer's disease, a range of infectious disorders including HIV, malaria, and other parasitoses [3].
Justification for a mixed approach
The utilisation of bivalent drug design is driven by the possibility of thermodynamically advantageous connection between bivalent ligands and distinct receptor recognition sites compared to monovalent ligands [4]. Such as with HIV protease receptors and steroid receptors, the bivalent ligands may also homo/homodimerize between two distinct receptors in adjacent proximity [5]. Bivalent ligands have dual receptor binding, linkers are absolutely important. Excellent selectivity and efficacy relative to the monomer can be obtained from well-constructed hybrids by because of their precisely length and flexibility.
Structural hybridization
Over the past 10 years, the literature has extensively used terms such "hybrid drug," "conjugate drug," and "codrug." The Greek idea of hybris [6] forms the basis for the name "hybrid". The combination of two or more physiologically active compounds that produces a new entity called the hybrid molecule defines hybrid drugs. As a result, the hybrid approach in drug design provides an alternative to conventional therapy as well as a substitute for the coformulation of several active drugs. The etymology is appropriate for the newly developed therapeutic entities that are referred to as "hybrid drugs," because the recently evolved molecular species ought to demonstrate supremacy and possess the characteristics of every component. On the other hand, structural hybridisation has objective to create the novel, more efficient single medicines preferably less harmful than the many drugs used for certain pharmacological purposes [7]. The word "codrug" mainly refers to hybrid molecules made of two either similar or different drugs joined by a cleavable connection. Hybrids covalently bound to a functional molecule are the "conjugates," from the Latin term conjugate, meaning to mix or match. These might operate as inhibitors (or activators) of a certain enzyme or as carrier molecules, therefore increasing the biological activity [6]. Hybrids, whether called codrug or drug conjugate, are always new molecular entities that combine the chemical constituents of multiple pharmaceuticals and bioactive compounds. Despite this, the pharmacokinetics and pharmacodynamics of these new compounds will always differ from the parent medications. In this review, all of these terms are used interchangeably: hybrid medicines [6].
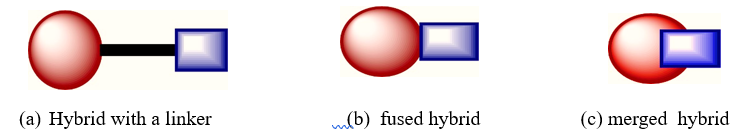
Fig. 1. Figurative representation of a hybrid featuring a linker (a), a fused hybrid (b) and a merged hybrid(c)
Linker architecture and partner selection
The hybrid drug may have two or more than two covalently bonded components, with or without a linker. Figures 1 and 3 depict the hybridisation mode, various permutations, and partner assembly combinations. It is possible to covalently combine the separate partners or molecular components into the molecular matrix in overlapping, fused, or linked configurations (Fig. 1) [8]. The topology of hybrid components can take several forms, such as a straight line, a branch, a cyclic structure, oligomers, polymers, symmetric, nonsymmetric, or branched. Molecular hybrid design isn't about coming up with a tonne of new designs; it's about getting better pharmacological effectiveness through selectivity and specificity. The goal of creating hybrids is to increase enhance pharmacological efficacy and reduce toxicity by innovative design. The partners in the hybrids should work together to improve the pharmacological activity that is desired. A complete understanding of the pros and cons of each drug or functional entity partnership should guide the hybrid design. Prior to commencing hybrid medication development, it is imperative to evaluate the unique biological targets of each partner, their proximity within the intracellular environment, and the pharmacokinetic and pharmacodynamic properties. Two or more drug molecules can undergo hybridisation, leading to the development of trimeric, dimeric and tetrameric compounds, together referred to as hybrid medicines. The choice of each medicine in a hybrid scaffold must be meticulously evaluated to provide complementary and synergistic pharmacological effects when integrating two or more different medications. Selecting two or more pharmacophoric components from various bioactive leads or drugs results in multifunctional hybrid molecules that are subsequently changed with appropriate functionality to enable the required coupling for hybridisation. The production of more comprehensive combinations is achieved by augmenting the quantity of active pharmaceuticals or pharmacophores incorporated into frameworks that combine chemicals. Although not the focus of this review, these are referred to as the multitarget drugs in literature. Hybrids involving two or three pharmacologically active compounds are the primary focus of this investigation because they are the most common and well-documented framework combinations that provide novel treatment opportunities. As shown by the situations of fused (Fig. 1b) and merged (chimeric) hybrids (Fig. 1c), a hybrid molecule could include a linker (Fig. 1a) or might not [9]. The components are assembled into a hybrid framework utilising a suitable attachment method decided upon by choice of a linker, in linked hybrids. The linkers should preferably be trifunctional or bifunctional cross-linked reactive molecules, or sterically unhindered ones. Incorporation of the different active chemicals into the hybrid scaffold depends on functionalisation. Dependent on the particular needs of the hybrid design, they might be dendritic, branched, or linear anchors with hydrophobic or hydrophilic properties. The linker moieties have mostly functional groups of alcohols, thiols (sulfhydryls), main amines, acids, or halides. The overall efficacy of the hybrid relies on its the location of the linker and its physicochemical characteristics; thus, it is preferable for the two components to be connected at a non-pharmacophoric location to preserve the integrity of the functional domains. Linkers are categorised as either non-cleavable (non-degradable) or cleavable (metabolically degradable) based on their drug release mechanism and in vivo stability [10].
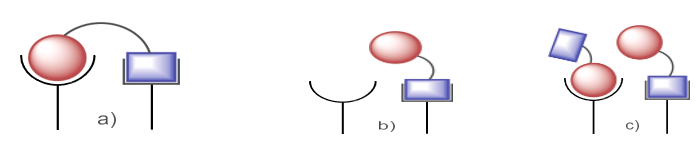
Fig. 2. (a) Parallel binding is the result of hybrid with sufficient linker lengths and nature.; (b) and (c) Parallel binding is not possible with insufficient linkers.
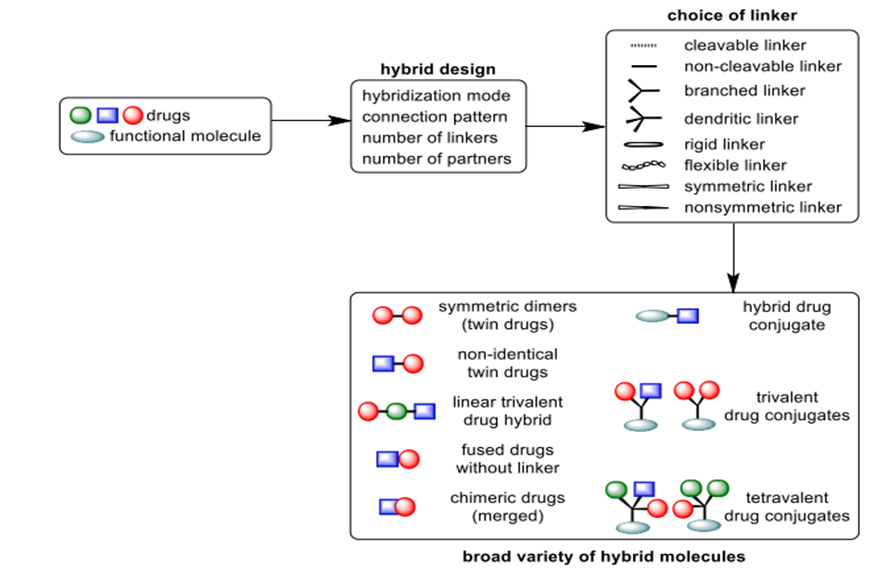
Fig. 3. A wide range of conjugates is produced through the molecular Lego construction of compound pharmaceuticals.
Linker types can be distinguished as either stiff, non-linear (such as aromatic, heteroaromatic, or bridging alicyclic) or flexible, linear, like methylenic or polymeric chains. Cleavable linkers start the breakdown of the hybrid by means of the pH, enzyme, and chemical concentration-related physiological milieu of metabolism, so releasing separate pharmacologically active entities for autonomous interaction with certain biological targets [11]. While enzyme-labile linkers, typically made of ester bonds, are susceptible to hydrolysis by plasma esterase, non-labile linkers, like ether or alkyl chains, can enhance activity by significantly shifting the hydrophobic-hydrophilic equilibrium of the conjugate. This, in turn, affects the compound's pharmacological and pharmacokinetic characteristics, and lastly its total effectiveness [12]. Other important factors, such as the linkers' length (spacer arm), flexibility, and conformational mobility, determine how well the hybrid's individual components interact with the two separate recognition sites that are close together inside the intracellular matrix or cell membrane [13]. Due to poor design and lack of information about the relative positions of the two targets within the cell or on the membrane, a hybrid that is too short and has a stiff linker cannot bind to both receptors at the same time (Fig. 2). While assembling all of the components, the mechanism of action of each ligand should take precedence. The linker has a big influence on how safe and effective hybrid medications due to the fact that its chemistry determines the release profile of the active ingredients [14]. Hybrids conjugated to functional molecules, symmetric di-/-trimers, and nonsymmetric di-/-trimers are the main types of drug hybrids shown in Figure 3.
Hybrid drug design through Lego-assembly of bioactive molecules
Traditionally a difficult, long-term, expensive procedure marked by a very high attrition rate, Lego's mixed design chemistry provides a captivating and dependable approach for drug development. Since it is a simple and pragmatic approach for synthesising unique bioactive chemicals with increased efficacy, the "Lego" chemistry concept for hybrid creation has great benefits [15]. Hybrids are synthesised using bioactive molecular building pieces that are easily accessible and may be assembled into a final structure using only a few straightforward reaction steps (Fig. 4). This method follows this principle. More thermodynamically favourable reactions have been identified, in addition to esterification and click chemistry, two widely used methods of hybrid assembly. Some examples of these reactions are cycloaddition, which opens the nucleophilic rings of epoxides and aziridines; hydrazone and heterocycle synthesis; adds to carbon-carbon multiple bonds; Michael additions; and non-aldol carbonyl reactions.
Potential combatants of drug resistance: Hybrid pharmaceuticals
Public health suffers a great risk from the rise of multidrug-resistant microorganisms. Unlike antifungal, antiviral, or antimalarial drugs, bacterial resistance to chemotherapy has been especially raised by the abuse of antibacterial drugs. According to current estimates, about 55,000 deaths yearly in Europe and the United States are caused by antimicrobial resistance (AMR). Global estimates of antimicrobial resistance (AMR) point to over one million deaths annually [16]. The pathogenesis linked with drug-resistant E. coli, S. aureus, K. pneumoniae, tuberculosis (TB), malaria (Plasmonium falciparum), and HIV has been identified as a possible threat to our survival in the future [17]. This is because drug resistance is getting worse and the number of infections is going up. Antimicrobial resistance (AMR) is going to be a big problem for health in the coming years because there aren't many drugs that can effectively fight infectious diseases caused by bacteria that are resistant. Hybrid drugs are one way to help solve the problem of drug tolerance, which is getting worse [18].
Antimalarial hybrids
From the beginning of civilisation, malaria has been a recurring disease of major public health concern. Millions of people worldwide still die from infections brought on by Plasmonella falciparum, the malaria parasite with the greatest death rates even although multiple antimalarial drugs are now under successful development. The antimalarial medications quinine and artemisinin are mainly derived from two plants: Artemisia annua L. and cinchona tree bark, respectively. They were the basis for the production of several powerful antimalarial drugs, including chloroquine, mefloquine, primaquine, mepacrine, dihydroartemisinin, artemether, artesunic acid, and artemisone [20].
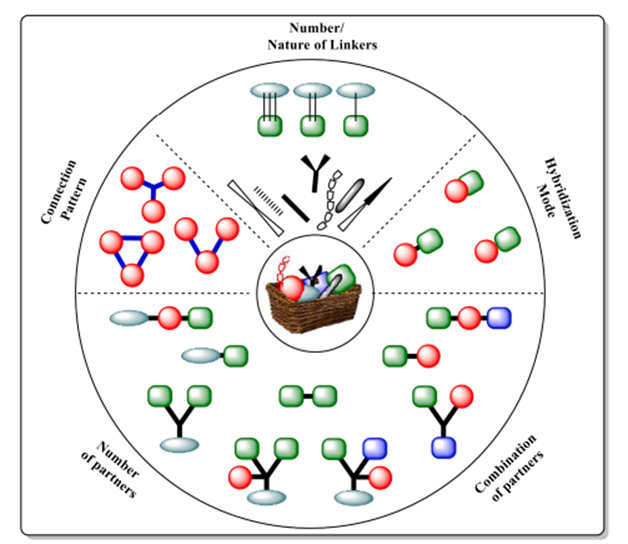
Fig. 4. A molecular Lego model for the creation of hybrid drugs; a matrix for hybridization
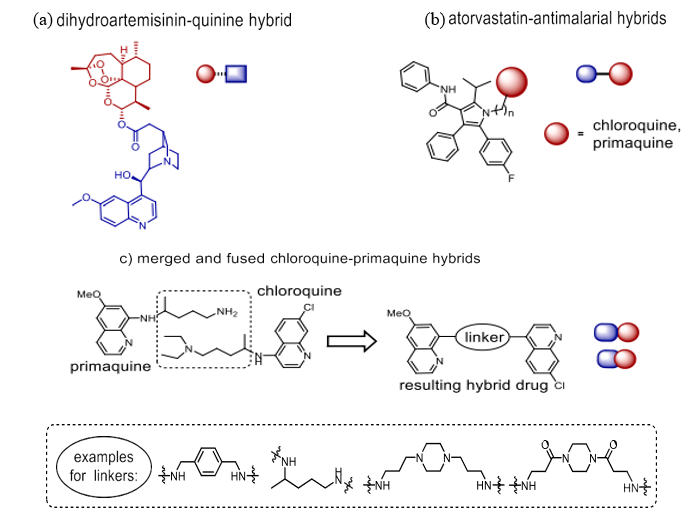
Scheme 1. Novel antimalarial drug-hybrids
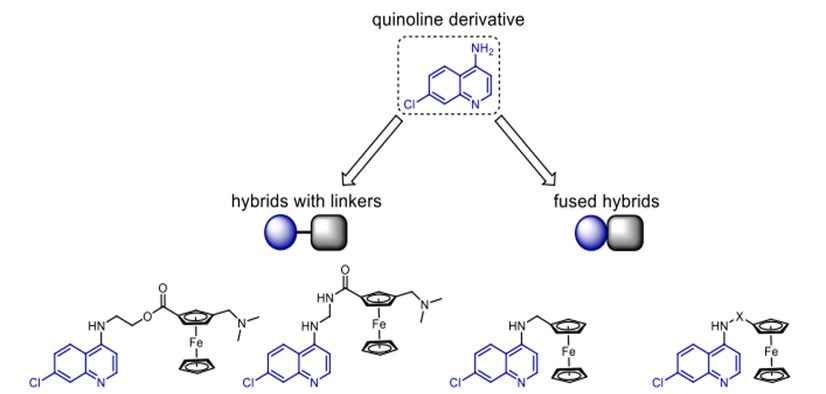
Scheme 2. Antimalarial compounds that are hybrids of quinoline and ferrocene.

Scheme 3. Redox-active Fe (II) activates intracellular artemisinin prodrugs.
Currently, Derivatives of artemisinin are the first-choice drugs for combination treatment of P. falciparum infections resistant to chloroquine [19]. The discovery Drug-resistant P. falciparum strains are a growing concern due to the parasites' ability to evade these medications globally, so increasing the urgency to find alternative antimalarial medications even if their pharmacokinetic parameters are favourable and their potential efficacy and safety profiles are promising. Emerging as a major breakthrough in the battle against multidrug-resistant P. falciparum infections is the hybrid medicine concept. A series of studies has been published that specifically address concerns of potency, toxicity, drug resistance, solubility, pharmacokinetics, metabolism, and delivery modalities, focussing on synthesis and antimalarial efficacy of drug hybrids. This book examines recent advancements in the field, specifically exploring the structural hybridisation of pharmaceuticals as a viable approach to developing effective antimalarial treatments. Connecting dihydroartemisinin to quinine's carboxylic acid derivative via an ester bond (Scheme 1a) Walsh et al. created a simple medication that can be broken apart to kill malaria parasites [23]. It was intended to address the issue of lipophilic artemisinin, which is rapidly flushed out of the body and causes great rates of regrowth when taken alone. Combining artemisinin with polar quinine derivatives was supposed to increase the half-life of the artemisinin molecule in living entities. Tests did not show this, but the combo medicine is less likely to grow resistant for these types of hybrids, where each component functions in its own right [24]. Against cultured P. falciparum, the hybrid is around three times more efficacious than either quinine or artemisinin alone or combined in a 1:1 ratio [23].
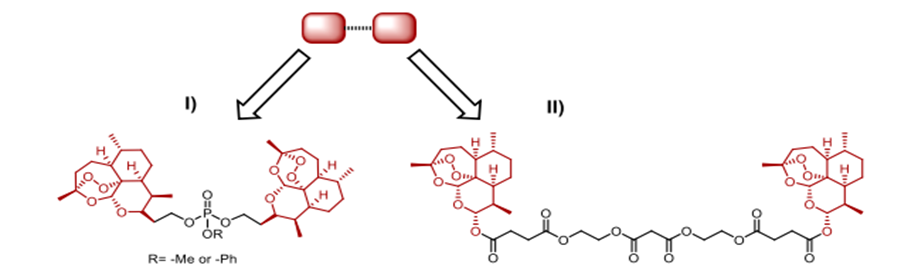
Scheme 4. Symmetric dimers with a cleavable linker that are based on trioxane and artesunic acid
Altering the binder between the two pharmaceuticals enabled Lödige et al. to establish a library of chloroquine-primaquine hybrids (Scheme 1c). In examining several phases of parasite life: notably the gametocyte stage (P. falciparum—most hybrids display higher effectiveness than their parent compounds), the blood stage (P. falciparum for 3D7/Dd2/K1 strains), and the liver stage (P. berghei). Mixed with many aminoquinoline antimalarials and artemisinin, a well-known HMG co-enzyme reductase inhibitor named atorvastatin performed better (Scheme 1b) [26]. Depending on the linker utilised, the atorvastatin hybrid including a tetra-methylene spacer showed an IC50 value between 0.40 and 1.41 µM [27].
Without altering how effectively chloroquine functions, ferrrocene prohibits it from functioning against P. falciparum. Many ferrocenyl hybrids of chloroquine have been synthesised during the past ten years using varying linkers to modify their action. Scheme 2 has several well-known members of this series. Whereas ferrocene-quinoline hybrids with stiff linkers had no impact, Flexible linkers allowed hybrids to be rather efficient against P. falciparum D10 and Dd2 strains [29]. Although artemisinin has great action against malaria as has already been noted, we shall discuss more specifically below. Studies have revealed that artemisinin's endoperoxide bridge is the primary structural element responsible for the action, even if the exact way artemisinin and its derivatives combat malaria parasites is not known at this time. Because their haemoglobin digestion is much improved, parasites produce a lot of Fe (II [30]). Iron (II) causes HO, H2O2, 1O2, and O2 to be released when it combines using artemisinin's endoperoxide unit. These radicals damage DNA, which kills parasite cells in turn (Scheme 3). By using an assortment of linkers and other dynamic pharmacophoric adjuvants, researchers were able to combat resistant P. falciparum strains with several artemisinin-based hybrid medications. There have been several reports of di-, tri-, and tetrameric hybrids with different sets of linkers derived from artemisinin analogues between 1999 and 2019. Olefinic, phosphate, bipiperidine carbamate, ether, and ferrocene are all possible examples. This discussion will focus on recently reported hybrids with significant activities from the aforementioned compound groups.
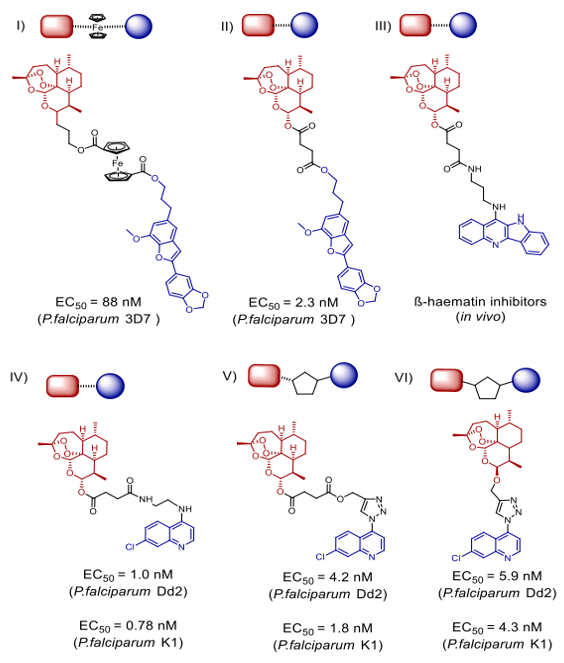
Scheme 5. Artemisinin-egonol-(indolo) quinoline antimalarial hybrids.32–34
Hybrid medications that are both symmetric and cleavable, connected by phosphate esters, were created by Jeyadevan et al. were featured in Scheme 4(I). In tests against tuberculosis (PCP) strain K1, which is resistant to chloroquine (IC50 = 0.2 nM) and chloroquine-sensitive strain HB3 (IC50 as low as 0.09 nM), these hybrids showed incredible antimalarial efficacy, with a 4700-fold increase compared to chloroquine and a 300-fold increase compared to artemisinin, respectively [31]. Scheme 4, II shows a symmetric 1,2,4-trioxane dimeric hybrid that outperformed dihydroartemisinin, artesunic acid, and chloroquine in terms of 1.4 nM EC50 and 3D7 distribution indicate antimalarial action. The artesunate-quinoline hybrid medications developed by Wang et al. outperformed their pharmacophores (Scheme 5, III) in terms of parasite killing efficacy [32]. The ?-hemolytic inhibitory effects were shown in live mouse models after four days of treatment at a dosage of 10 mg/kg daily. The result was a marked decrease in parasitemia and an average mouse lifespan of 7.7 days. From dihydroartemisinin, 1,2,4 trioxane, artesunic acid, and useful natural compounds like egonol or homoegonol, our group developed a variety of fused hybrids (cleavable and non-cleavable) (Scheme 5, I and II). The combination of medications effectively inhibited the parasite's maturation in Plasmodium falciparum 3D7 infected red blood cells (RBCs) (EC50 in nanomolar concentration) [33].
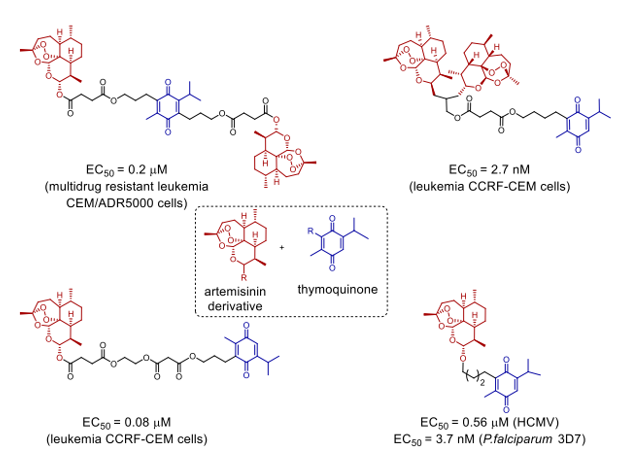
Scheme 6. Orthogonal construction artemisinin/artes unic acid and thymoquinone have shown a novel family of hybrid molecules having anti-cancer, anti-viral, and antimalarial properties.35
Novel artemisin-quinoline hybrids (Scheme 5, IV-VI) have recently been synthesised, and they have demonstrated promising effectiveness against P. falciparum strains K1 and Dd2 resistant to chloroquine and multidrugs. Whereas for chloroquine the EC50 values were 165.3 nM and 302.8 nM, respectively, the values for Dd2/K1 were as low as 1.0 nM and 0.78 nM. The hybrids are likewise drastically lowering the experimental malaria parasitemia.thirty-three CuCAA, a method based on copper as a catalyst, allows one to readily synthesise the new compounds by amide bond production. Chemical proteomics [34] allowed successful identification of possible artemisinin-quinoline hybrid-binding protein targets. Killing P. falciparum 3D7 (with an EC50 as low as 3.7 nM), HCMV (with an EC50 as low as 0.56 µM), and anticancer cells the great possibility of hybrid synthesis is shown by the great variety of activities presented by the hybrid compounds given the diverse range of biological functions exhibited by artemisinin and its analogues [35].
Antibacterial hybrids
One major problem with antibiotics is that they can be overused, which leads to the rise of bacteria that bacteria that are resistant to antibiotics of both the gram-positive and gram-negative kind. There is growing concern about bacteria that are resistant to different treatments, specifically ESKAPE, which stands for Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species as a whole [36]. The existing antibiotics are ineffective against the gram-negative bacteria due to its two-layer protection and strongly expressed efflux pumps. Of the many potential responses to this issue, drug hybrids are the most significant. Numerous antibiotic hybrids are currently being tested in human clinical trials. An interesting new path in the battle against the drug-resistant bacteria and for raising the efficacy of the previously available antibiotics is the development of antibiotic hybrids [37]. Here we shall review the most recent advancements in antibiotic hybrid technologies together with challenges.
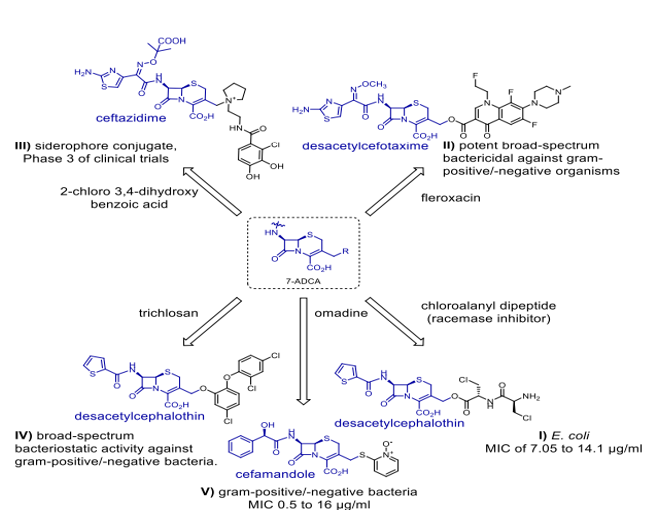
Scheme 7. Hybrids of antibiotics from 7-ADCA (7-aminocephalosporanic acid).
Omadine is a cephalosporin derivative coupled with a non-?-lactam antibiotic, is a recognised inhibitor of bacterial ARP production [38], making it the first hybrid efficient against gram-positive as well as gram-negative bacteria. Still, the unpleasant systemic toxicity of omadine made this combination useless therapeutively. Scheme 7 (I) shows desacetylcephalothin in relation with a chloroalanyl dipeptide, which acts as an inhibitor of alanine racemase [39]. The hybrid shows activities directed against E. coli. Initiated for several composite entities based on cephalosporins were preclinical and clinical studies. NewBiotics Inc.'s NB2001 (Scheme 7, IV) exhibits potent effectively inhibits the growth of gram-positive and gram-negative bacteria across a wide range of the utilisation of a desacetylcephalothin linked with triclosan. The in vivo release of triclosan was responsible for the action of NB2001. Desacetylcefotaxime (Scheme 7, II) is covalently bonded to fleroxacin by a cleavable ester linkage in the hybrid prodrug, demonstrating significant effective against a wide variety of bacteria, including gram-positive and gram-negative [41]. The hybrid demonstrates antibacterial efficacy against E. coli isolates cannot be killed by either ?-lactams or both, with NB2001's action derived from the in vivo release mechanism of triclosan. A cleavable ester link in the hybrid prodrug chemically
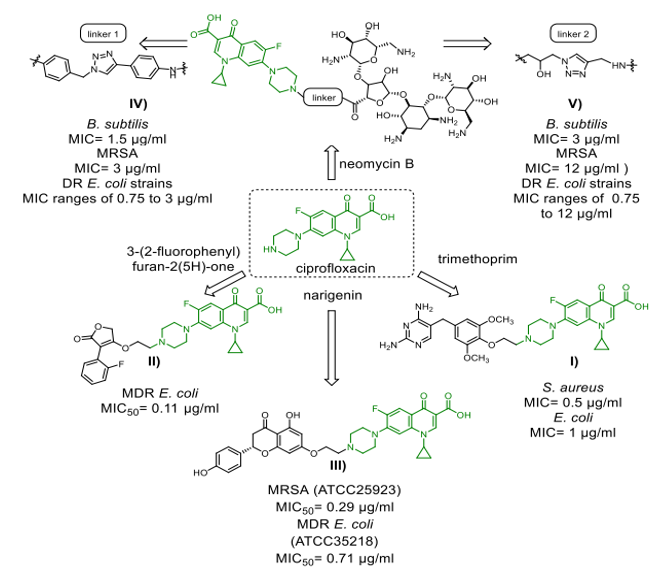
Scheme 8. Ciprofloxacin-derived antibacterial compounds
bonds transitioning from desacetylcefotaxime (Scheme 7, II) to fleroxacin, a powerful bactericidal agent that kills both gram-positive and gram-negative bacteria,[41]. Exhibiting antibacterial effectiveness in E. coli isolates Enzymatic hydrolysis of the ?-lactam ring structure destroys the hybrid in vivo, regardless of whether it is resistant to ?-lactams alone or both ?-lactams and fluoroquinolones [41]. Additionally, the hybrid was found to decrease proinflammatory cytokine production while simultaneously increasing LPS-stimulated tumour necrosis factor alpha (TNF-a) induction.42 Siderophores and other hybrids trick bacteria into taking more medication by using their iron transport channels [43]. Presently in preclinical or clinical research are many ?-lactam siderophore hybrid drugs. By seizing the bacterial iron transport system, the siderophore—an iron-chelating adjuvant—has greatly raised intracellular drug concentrations. Especially remarkable in this range are the cephalosporin-catechol hybrid cefiderocols (Scheme 7, III), which are under phase 3 clinical studies [43]. Most often consisting of a ciprofloxacin unit, fluoroquinolone pharmacophore is found in the vast majority of antibiotic-hybrid medications that have lately been published and are active against gram-negative bacteria. As well as their well-established structure-activity correlations (SAR), fluoroquinoline antibiotics' structural resilience contrasts with the restricted time period during which ?-lactam medicines remain chemically stable makes them perfect candidates for hybrid creation [44]. Hybrid antibiotics that combat gram-negative infections come in a variety of forms, including cleavable linkers and non-cleavable fused hybrids (as seen in Scheme 8). More effective than 5.23 µM for ciprofloxacin in inhibiting DNA gyrase in vitro was TyrRS inhibitor 3-(2-fluorophenyl) furan-2(5H)-one, which is is associated with ciprofloxacin (Scheme 8, II). This drug likely has a twofold mode of action because the hybrid medicine has comparable in vitro TyrRS-inhibitory efficacy to the famous 3-arylfuran-2(5H)-one-based TyrRS inhibitors [45]. In addition, two neomycin B-ciprofloxacin instances are displayed. Two hybrids, shown in Schemes 8, IV, and V, include an aromatic triazole connector and a hydroxyl group functionalised 1,2,3-triazole linker. The neomycin B-ciprofloxacin hybrids may have an effect on gram-positive Bacillus subtilis, gram-negative drug-resistant Escherichia coli, and methicillin-resistant Staphylococcus aureus (MRSA). Apart from these instances, Scheme 8, III shows the flavonoid naringenin-linked ciprofloxacin as is Trimethoprim, which is attached to the piperazine ring of ciprofloxacin-BP4Q-002, [47] and shows strong efficacy against S. aureus. The hybrid P. aeriginosa is a prevalent opportunistic pathogen that causes nonsomial infections in individuals with compromised immune systems. It exhibits antimicrobial efficacy against methicillin-resistant Staphylococcus aureus, gram-negative bacteria, and fungus (amphotericin B-resistant Candida albicans). The World Health Organisation (WHO) has published a list of the most hazardous superbugs, which includes carbopenam-resistant P. aeruginosa. It is now evident that hybrid medications that are based on tobramycin can outperform conventional antibiotics. Scheme 9 illustrates numerous intriguing hybrids that demonstrate efficacy against resistant P. aeruginosa. Every hybrid that has been demonstrated is composed of non-cleavable pliable connectors that are composed of a 12-C atom bridge. These hybrids combine an antibiotic like moxifloxacin, ciprofloxacin, or lysine-peptoid with an efflux pump inhibitor (EPI) like paroxetine, 1-(1-naphthylmethyl-) piperazine (NMP), or the aminoquinoline analogue D-Ala-D-hPhe, which is similar to the dibasic peptide (DBP). Apart from their resistance to hybrid medicines (Scheme 9) have activity against several bacteria, such as P. aeruginosa [48], S. aureus, E. coli, and MRSA (Methicillin-resistant Staphylococcus aureus).
Anticancer hybrids
Approximately 1.9 million new cases were reported in 2016, with 0.7 million fatalities[49], cancer remains the most common cause of death in the United States despite the amazing developments in anticancer therapy throughout the previous two decades. As scientists look at other, safe, and efficient cancer therapies, the hybridisation idea has grown ever more important. Many anticancer medications have been hybridised from synthetic pharmaceuticals and natural items thus far to assess their efficiency against several cancer types. Several studies that provide a basis for hybrid design have been published in this sector [50]. There is a history of successful usage of a wide range of structural classes in the semisynthetic synthesis of hybrid compounds. These structural classes include steroids, polyphenols, alkaloids terpionoids, and anthracyclics. Furthermore used in the composite design were many non-natural pharmacophores, including acridone, vorinostat, bexaroten, and gefitinib. Like other drug categories, various linker methods (cleavable and non-cleavable), have been used to create creative anticancer hybrids fit to the particular cancer targets. Developed by Pfizer, a C-17 ?-phosphate ester of estramustine has previously been used clinically for prostate cancer therapy [51]. Apart from this example, numerous more hybrid anticancer prospects have proceeded to their phase II and III of their clinical trials. This portion highlights anticancer hybrids generated
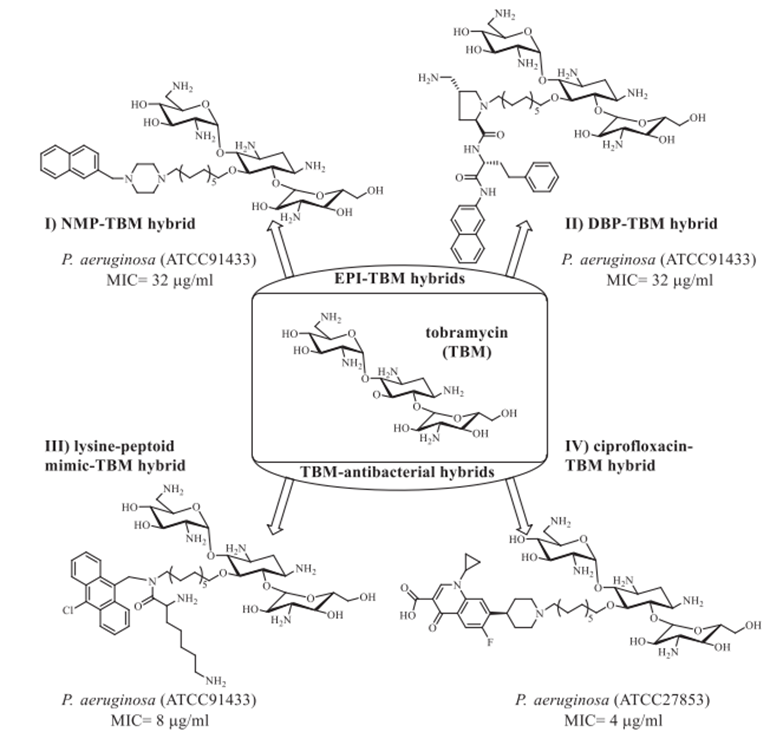
Scheme 9. Effective tobramycin hybrids against Pseudomonas aeruginosa.48
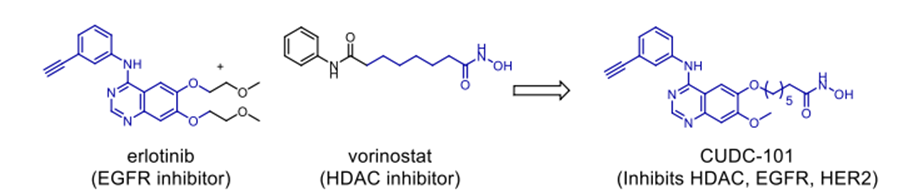
Scheme 10. Anticancer hybrid CUDC-101.
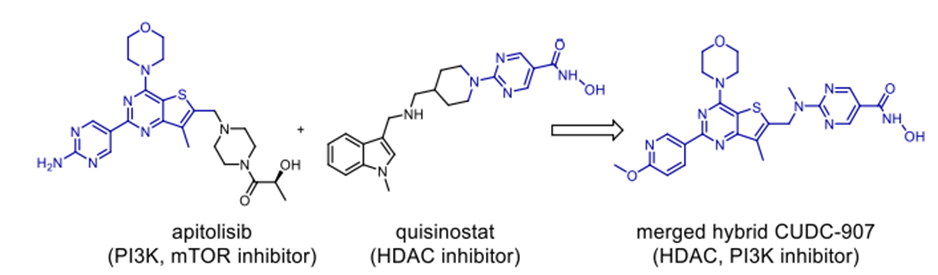
Scheme 11. Anticancer hybrid CUDC-907
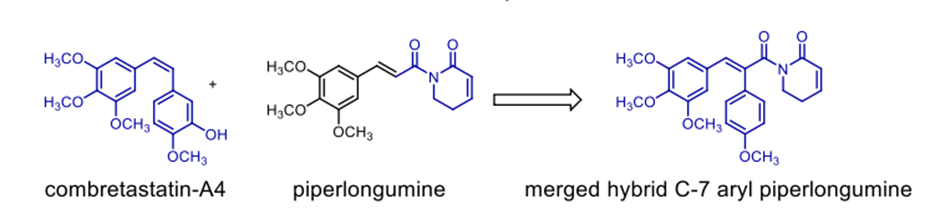
Scheme 12. C-7 aryl piperlongumine, an anticancer hybrid
lately and demonstrating considerable efficacy. FDA-approved treatments clinically used to treat cutaneous T-cell lymphoma (CTCL) and non-small-cell lung cancer (NSCLC) respectively are Vorinostat and erlotinib. In response to the resistance metastatic cancers have evolved against both treatments, scientists have created a merged hybrid combining the two medications. Produced successfully, the hybrid CUDC101 efficiently suppresses HER-2, EGFR, and HDAC [52]. Against advanced head, neck, gastrointestinal, breast, liver, and solid tumours (Scheme 10), the clinical phase I trial of CUDC-101 has been essentially finished. Widely known as a hydroxamic acid, quinostat has proven to be able to block histone deacetylase (HDAC [54]). Apitolisib, a preclinially established inhibitor of mTOR and PI3-kinase, was added to the HDAC inhibitor. Both hybrid components exhibit five-ty oral action. CUDC-907, the combined hybrid of these two pharmacophores, reduces PI3K as well as pan HDAC (see Scheme 11). Blocking the PI3K-AKT-motor signal transduction pathways will help the hybrid chemical show antineoplastic action. Combretastatin-A4, which is documented to target colchicine binding sites, functions as a tubulin polymerisation inhibitor. 1957 57 Piperlongumine, an alkaloid derived from piperlongum, is a pro-apoptotic compound having cytotoxic effects on many human cancer cell lines [58]. Punganuru et al. synthesised a combination of these two powerful compounds, C-7 aryl piperlongumine chemicals, in Scheme 12, which demonstrates significant disruption of tubulin polymerisation and reactivation of the P53 mutation [59]. Many hybrids anticancer drugs have been developed from anticancer natural ingredients recently. Because of its adaptable anticancer action and somewhat low mammalian toxicity, Artemisinin has attracted special interest. Lombard et al [60] for instance created a symmetric aminoquinoline-linked hybrid. GI50 values within the 0.03 µM (MCF) to 0.08 µM (TK10) range [74] are shown in Fig. 5, I. Comparatively to etoposide [60], the hybrid shows a 74-fold increase in anticancer efficacy. Likewise, with an IC50 of 0.07 µM, the ferrocene-contacting dimer (Fig. 5, VIII) produced showed a sixfold increase in activity against leukaemia CCRF-CEM cells, in contrast to the parent chemical dihydroartemisinin (IC50 of 0.45 µM). With a 17-fold increase in activity as compared to the commercially utilised medication paclitaxel [62], the cytotoxicity of nonsymmetric dimer IV (Fig. 5) against BT49 cell lines was moderate to very excellent (IC50 up to 0.08 µM). In HL60 cells, dimer V, which is connected to phosphate, displayed the highest cytotoxicity (Fig. 5), with a toxicity level almost fourteen times greater than dihydroartemisinin (0.05 µM vs 0.71 µM). [63] Reference 64 indicates that the artemisinin hybrid including a sulphur bridge (VIII, Fig. 5) exhibited the highest potency against MCF-7, with an IC50 value of 0.025 µM. The guanidine dimer derived from artemisinin exhibited significant cytotoxicity across three distinct IC50 values for cancer cell lines varied from 0.24 to 1.42 µM [65]. Leukaemia cells are particularly vulnerable to the ferrocenyl hybrid's cytotoxicity (CCRF-CEM, IC50 = 10 nM) [61,66], as seen in Figure 5 (II). The Trioxane-Ferrocene hybrid showed a highly efficient cytotoxicity (IC50 0.57 ± 0.22 µM) against CEM/ADR5000 cells, which are multidrug-resistant leukaemia (Fig. 5, III) [75]. Schema 13 shows that a combination of vemurafenib and PAC-14028, also called pexidartinib, stops the growth of cancer cells by blocking the activity of the stem-cell factor receptor, the colony-stimulating factor-1 receptor (CSF1R), and FMS-like tyrosine kinase 3 (FLT3). Phase II clinical studies for recurrent glioblastoma and advanced castration-resistant prostate cancer were successfully completed by pediartinib [67]. The anticancer medication vinblastine and the folic acid molecule form a hybrid folate receptor ligand called vintafolide. The two are linked by a self-immolative disulphide linker and a hydrophilic peptide spacer (Fig. 6). In contrast to the cytotoxic and self-immolative linkers, which are required for the action, the peptide spacer can be changed without drastically affecting the activity. Phase II clinical studies for non-small cell lung carcinoma and platinum-resistant ovarian cancer were successfully completed by the hybrid molecule [68].
Antiviral Hybrids
Compared to other antiinfective hybrids, the literature has quite few reports on antiviral hybrids. Kimpe et al. [69] and Sasragiotto et al. [70] created two fused antiviral hybrids shown in Scheme 14. Purine ?lactams form the first hybrid; ?-carboline 4-thiazolidinones form the second hybrid. Furthermore, shown in Scheme 14, III are the newly discovered inhibitors by Jadav et al. [71]. We created the hybrids by combining MO2/DO3 with the ?-OG pocket of the DENV2 E protein that is involved in pharmacophoric interactions. This pocket is a possible target for the development of antiviral medicines that prevent the virus from entering the host cell. Out of the 44 merging hybrids created by these combinations, one demonstrated an EC50 value in the low micromolar range (1.33 ± 0.42 µM) when tested against DENV serotype-II [71]. Our lab has recently created one-pot method that does not include metals synthesise a targeted library of novel antiviral quinazolines that are 4,5,7,8-substituted. A novel class of artesunic acid-quinazoline hybrid molecules (I) (Scheme 15) [72] was constructed using the most effective compounds in this class. A strong antiviral effect against human cytomegalovirus (HCMV) was observed in the fluorescent hybrids that were produced. This medicine outperforms both quinazolines and artesunic acid, which are the parent compounds, and ganciclovir, the reference medication, with an EC50 value of 2.6 ± 0.5 ?M, with an EC50 as low as 0.1 ± 0.0 ?M. Similarly, our group's extremely cleavable dimeric (II) and trimeric (III) artemisinin hybrids shown remarkable antiviral properties with an EC50 ranging from 0.04-0.31 µM when tested against HCMV (Scheme 15) [22,73].
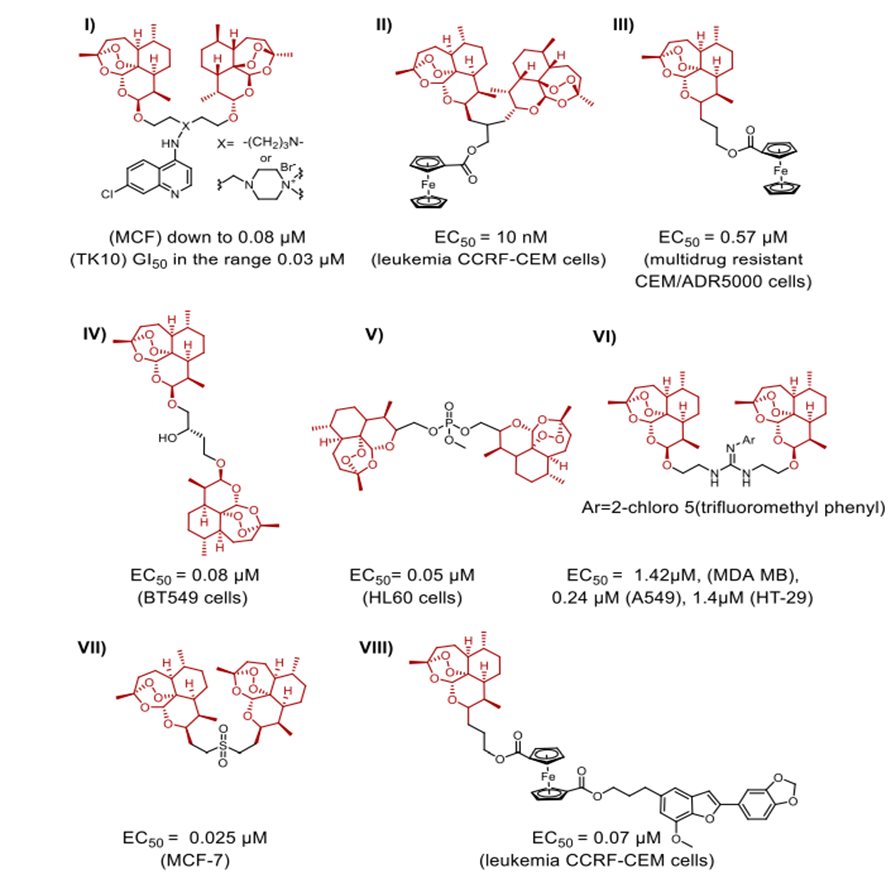
Fig. 5. Hybrids generated from artemisinin that exhibit strong anticancer effects
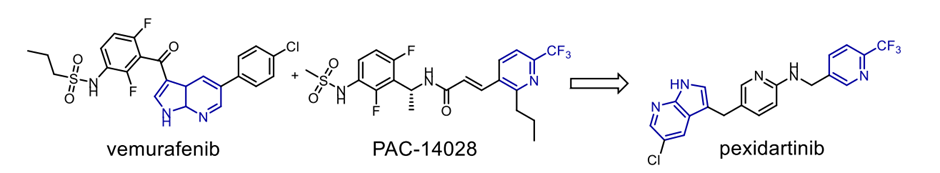
Scheme 13. Merged hybrid pexidartinib.
CONCLUSION AND OUTLOOK
A potential tool for the evolution of new drug discovery strategies, hybrid molecule design has advanced significantly during the past two decades. Novel, strong anticancer, antiviral, and antibacterial drugs developed thus already make use of the hybridisation idea. The chemistry involved in creating hybrid medications is typically rather straightforward, involving either the direct conjugation of two monomeric moieties that are similar or distinct, or the use of generic ligation procedures to ligate the hybrid's various parts with the appropriate designed spacer arm. The hybrids were created using a combination of cleavable and non-cleavable linkers. Linker design was shown to be quite important for the control of hybrid drug action. Linkers may serve as more than just spacers, facilitating the efficient interaction of individual hybrid components with biological targets, according to research into a variety of symmetric and nonsymmetric linkers. Moreover highly crucial for the pharmacokinetic and pharmacodynamic aspects are those elements influencing the general toxicity and efficacy of the drug candidate. Despite significant advancements in the last 20 years in the development of novel hybrid medications for the treatment of some cancers, viruses, bacteria, and malaria, there are still unanswered questions. Additional research is necessary to comprehend the structure-activity relationships (SAR) of hybrid medicines in order to facilitate linker design. We must ensure that the linker design can contextualise the hybrids' pharmacokinetic/dynamic properties and their broad pharmacological and toxicological effects.
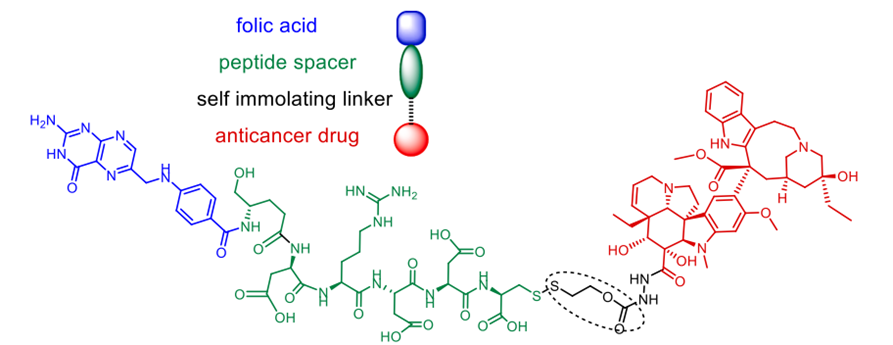
Fig. 6. An EC145 folate receptor ligand combination called vintafolide
It is critical to do research on the optimal spacer arm length, confirmation, and mobility in relation to the intracellular drug targets. The exact calculation of the spacer arm length is critical for all conjugates, but especially for those with a non-cleavable linker, as it allows the ligands to bind well to their corresponding biological targets.
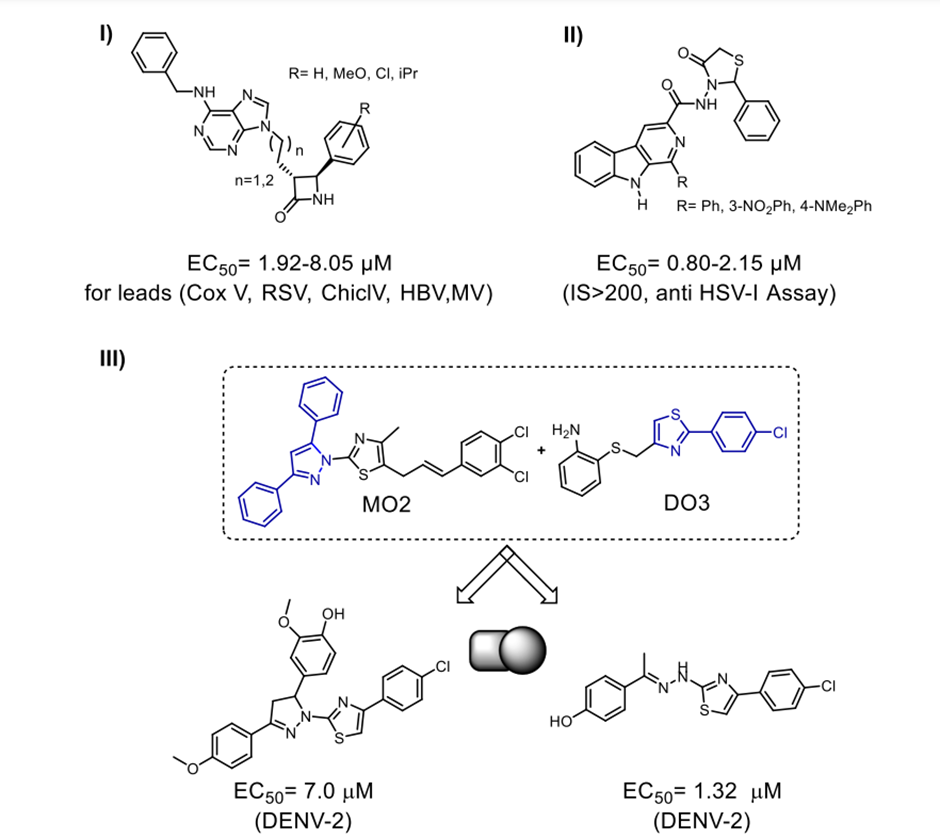
Scheme 14. I) compounds combining purines with ?-lactams; II) compounds combining ?-carboline with thiazolidinones; and III) compounds combining MO2 and DO3 to fight DENV-2. Hybrids fighting viruses.
Investigating further important factors such their therapeutic window, shelf life, formulation difficulties, and effect on drug resistance can help one to understand relevance of drug hybrids for their clinical uses. Ensuring that the final design is in line with the molecular weight and solubility characteristics in order to achieve drug-like properties with a suitable number of locations that receive or donate hydrogen bonds for efficient target binding presents the most important design difficulty to overcome when building drug hybrids. The most recent developments in this field must be fully exploited to grasp the changed efficacy of the hybrids from the perspective of the ligand-target interaction since the mechanistic studies concerning the enhanced efficacy of some hybrid molecules have not so far given a complete knowledge. Finally, compared to monotherapy, hybrid technology has become a powerful tool for the identification of more effective, benign drugs with great degree of specificity. Using the linker architecture, exploring new designs and further fine tuning with a suitable choice of collaborators and operational entities would help to find new drug candidates as safer and more effective substitutes for some current antiinfective and anticancer drugs. Since several hybrid compounds have commenced the early and late stages of clinical development, the idea of hybridisation as a new drug discovery approach offers great promise.
REFRENCES



Amar Kumar Mishra*, Kartikay Prakash, Nancy Tiwari, Structural Hybridization as An Efficient Method for Novel Drug Candidates, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 2, 1683-1704. https://doi.org/10.5281/zenodo.14898411
 10.5281/zenodo.14898411
10.5281/zenodo.14898411
