Brilliant Grammar School Educational Society Group of Institutions Integrated Campus - Faculty of Pharmacy.
Traditional drug development is a time-consuming process of target discovery (pathologies), compound screening through massive libraries and hit optimization which has facilitated many therapeutic successes over time. Yet traditional drug development fails to adequately model the intricacies of molecular interactions and subsequently becomes inefficient, compounding adverse events and failing to recognize what would otherwise be game-changing drugs. For example, steps like molecular dynamics simulations to predict what drugs will do before they're made can minimize the need for hit optimization and the trial and error that comes with it. Instead, step-by-step quantum simulations based on superposition, entanglement and quantum tunneling that gauge potential reactions at the molecular level can evaluate interactions before they occur to assess in what ways lab work can be minimized. For instance, quantum computing facilitates molecular docking, virtual screening, and molecular dynamics that factor in protein flexibility (which may differ between research), solvation effects (intermolecular and intramolecular interactions in solutions), binding free energy and reaction coordinates. Furthermore, respective methods including wavefunction simulation, quantum phase estimation, quantum walks and Monte Carlo methods, quantum tunneling and excited-state calculations allow a researcher to assess probabilities of positions, transitions, determinations of divergences and convergences, thermodynamics and spatial/temporal transitions of drug responses. Thus, with quantum computing and quantum machine learning capabilities, candidate drugs can be found and optimized faster with predictive qualities that not only speed up but facilitate cost-effective and accurate drug production while simultaneously opening more chemical spaces for exploration. As quantum technology continues to become more advanced, pharmaceutical R&D will become more accessible, effective and efficient for both researchers and patients.
The?????? traditional drug discovery avenue is a very lengthy and expensive way. It starts from a target (a molecule associated with a known disease). Then researchers are forced to check through millions and millions of chemical libraries or computer models to find a hit (molecules that can bind to the target). In the following stage of development, hit-to-lead, the hits are tested for power and lessened side effects and, therefore, bettered for uptake and release. Even though this step-wise method has produced millions of successful drugs, it is still a very time-consuming and costly ??????undertaking. One?????? of the major drawbacks of traditional drug discovery methods is their failure to portray intricate molecular interactions realistically. Conventional computational methods usually have a hard time figuring out the binding of drug candidates to their targets, thus throwing up ineffective compounds, side effects not considered, and opportunities for discovering new therapeutics going unnoticed. Introducing quantum simulations in drug discovery is a promising solution to these problems as they use the principles of quantum mechanics, such as superposition and entanglement, to their advantage. The quantum-level modelling of molecular interactions by quantum simulations can thus make drug discovery a lot faster, the different experimental trials few, and the predictive reliability high. Their use, which is first of all proposed for large molecules, is also envisaged for small molecules hence several pharmaceutical research applications are ??????possible. Quantum?????? simulations is a technique that can give drug discovery several advantages. They allow for accurate modelling of molecular interactions, and also can be used to give detailed insight to the chemical reactions and the biological processes such as enzyme activity or protein folding. Using quantum simulations, the properties of drug molecules can be predicted which is very helpful in selecting the molecules that are most likely to be effective as drugs. They limit the need for extensive laboratory testing because they can identify the most promising compounds for synthesis and evaluation. Besides that, quantum simulations enable personalized medicine by predicting how a given drug will interact with an individual biological profile. All these capabilities combined are a powerful tool in the hands of researchers and they promise to revolutionize drug discovery making it much more precise, faster, less expensive and still deeply understandable complex molecular ??????systems. Molecular?????? docking is an instrumental technique in drug design through which it explains the binding of ligands, i.e., small molecules, with proteins - receptors of the body - resulting in stable complexes. In the present era, it is highly relied on different advanced computational tools such as computer-aided drug design (CADD), quantitative structure-activity relationships (QSAR), and deep docking methods. With the help of these technologies, vast molecular libraries can be screened in a very short time leading to identifying the best drug candidates. Besides, quantum computing is enabling the scientists to refine the protein models in terms of flexibility, solvation, and quantum mechanical interactions while at the same time making the studies of large biomolecular corelations much faster. Virtual screening is another indispensable tool in drug discovery which entails the screening of extensive chemical libraries to pick up the most lead compounds. Conventional methods are hindered by factors such as simplification of models, computational power requirements, and the challenges of exploring vast chemical spaces. Quantum machine learning (QML), which is a combination of quantum computing and machine learning, can overcome these limitations by proficiently representing molecular features, understanding complex quantum patterns, and finally speeding up the process of the most potent drug candidates' identification. Some of the quantum-enhanced techniques that include quantum simulations, quantum neural networks, quantum kernels, and quantum search algorithms, facilitate virtual screening to be done faster, with higher accuracy, and in a more efficient way besides cost savings and experimental workload reduction. Molecular dynamics (MD) simulations are crucial tools in drug discovery depicting the movements and interactions of molecules during a certain time period. Classical MD regularly runs into problems when asked to handle big or complicated biomolecular systems because of limits to computational power. This is where quantum algorithms can help by converting the problems into quantum ones and thereby achieving higher precision in calculating binding energies, representing conformational changes in proteins and even modeling solvent interactions. Besides that, quantum-enhanced MD techniques such as wavefunction simulations, quantum phase estimation, quantum walks, quantum Monte Carlo methods, and excited-state calculations can be used to understand molecular behavior, reaction pathways, thermodynamics, and electronic transitions in detail. Altogether, these strategies can result in quick, accurate, and even predictive models of drug-target interactions which is a huge step towards drug discovery monetarily and otherwise. Quantum simulations and quantum-enhanced computational methods represent a paradigm shift in the arena of drug discovery. By adhering to quantum mechanics principles and combining them with innovative computational techniques, they not only allow to model molecular interactions with utmost accuracy but also considerably accelerate the processes of identifying and refining drug candidates aimed at personalized medicine and novel therapeutics. In due course, as quantum computing technology advances, its use in drug discovery would be a major factor in improving the efficiency, effectiveness and scope of pharmaceutical development ??????research.
QUANTUM SIMULATIONS IN DRUG DISCOVERY
Typically,?????? drug discovery begins with the identification of a molecule associated with a disease, referred to as the target. Scientists explore vast chemical libraries or use computational models to find compounds that can bind to the target. Such compounds are called hits. These hits are then refined in the hit-to-lead stage to enhance the effectiveness, reduce the side effects, and ensure that the drug is absorbed and eliminated properly in the body. Although this multi-stage procedure has led to the development of numerous drugs, it is still very time-consuming and ??????costly.
One?????? of the biggest hurdles for traditional drug discovery is that it is difficult to replicate accurately how molecules interact within the body. Therefore, in some cases, scientists make wrong predictions of how drugs will bind to their targets, thus incurring a loss of time, unexpected side effects, or the overlooking of new treatments that could be effective. A quantum simulation to be a promising answer to this problem.
Using advanced quantum computing (QC) algorithms, scientists are able to simulate such interactions to a very high degree of accuracy. To the extent that they make use of the quantum computers features such as superposition and entanglement, researchers will be able to anticipate drug behaviour, thus the process of discovering will be faster and fewer will be the experiments that need to be conducted. The advantages that come from this technology are mainly for complex molecules that are large, but even small molecules can benefit from quantum ??????simulations.
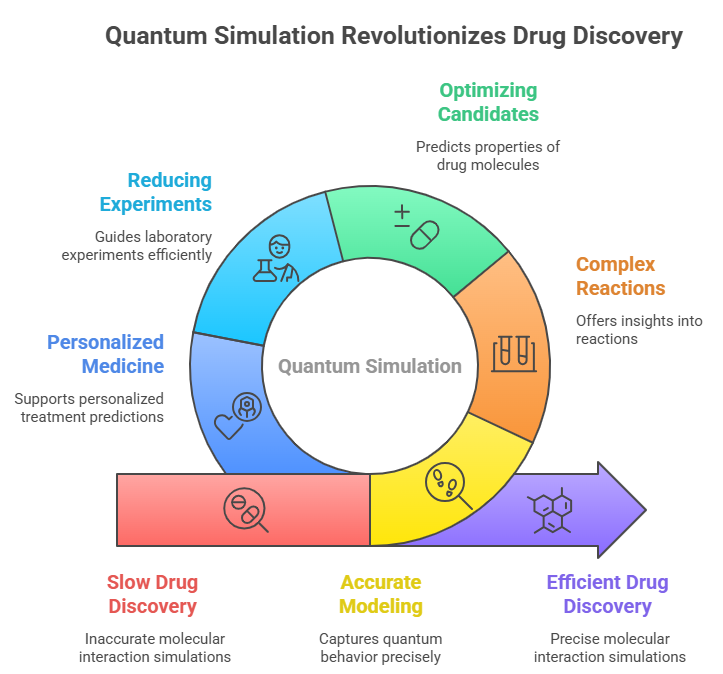
Fig 1. Quantum Simulation Revolutionizes Drug Discovery
Accurate?????? Modeling: Quantum simulation is a direct reflection of the quantum nature of molecules, thus, it allows for more precise predictions of their interactions among themselves and with biological entities [2].
Understanding Complex Reactions: With the help of quantum simulation, one can delve into the minutest details of chemical reactions and biological processes that constitute drug development, such as enzyme interactions and protein folding [3].
Optimizing Drug Candidates: Besides, in advanced quantum simulations, the molecular structures can be predicated, thus, the researchers will be led on a promising path to the therapeutic effect [4].
Reducing Experimental Efforts: Quantum simulation can be a key player in the facilitation of highly targeted laboratory experiments by determining which compounds for synthesis and testing are most likely to yield results [5].
Personalized medicine: One of the ways quantum simulations can be of great help is in the personalization of treatments as they have the ability to predict drug interaction with the individualized biological profile ??????[6].
Briefly,?????? Quantum simulation might be the means by which the idea of a paradigm change in drug discovery is brought to reality, giving not only a more accurate way but also one that is inherently efficient in modeling and understanding complex molecular interactions[7]. Consequently, a technology such as quantum computing, even if it is still quite far from being practical and ready for use, could be a massive drag that really accelerates the development of next-generation drugs and therapies. These sections explain the steps involved in drug discovery simulations and also how quantum computing can be utilized to accomplish them ??????better.
MOLECULAR DOCKING AND QUANTUM COMPUTORS
It?????? is essential in molecular biology and drug design to predict interactions between proteins and small molecules, which are called ligands. Molecular docking stands out as a popular computational technique to achieve these goals. Docking determines how ligands fit the binding sites of proteins, thus giving the most binding orientations and conformations. Consequently, the scientist is able to predict the ligand binding affinity and potential biological activity.
The most advanced molecular docking also features elaborate computations like computer-assisted drug design (CADD), quantitative structure-activity relationships (QSAR) and deep docking methods to generate a detailed interaction scenario between small molecule and protein target. As a result, it is possible to screen huge molecular libraries in a fraction of the time, thus drug development can be done with only the best candidate molecules. This simplified example is a demonstration of how molecular docking is a powerful tool to micro-dose the drug discovery journey.
Normally, the procedure begins with selecting molecules from large chemical libraries which are then subjected to CADD. CADD is intended for locating the best candidate drugs with good pharmacological properties in a very short time and at the same time it indirectly reduces experimental testing in terms of cost and time. It is a blend of diverse computational methods like molecular modeling, virtual screening, and molecular dynamics simulations. CADD techniques can be either structure-based or ligand-based. In ligand-based methods, small molecule-protein interactions are studied with the help of quantum algorithms that assist drug design optimization, whereas in structure-based methods quantum models are used to predict biomolecular structures and thus facilitate the identification of targets. Quantum computing is expected to revolutionize drug discovery by making it not only accurate but also efficient ??????Fig.2.
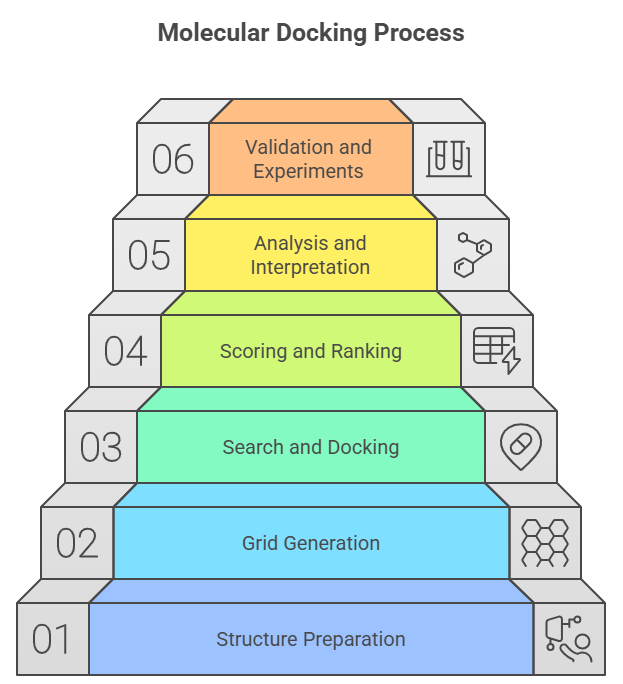
Fig 2. Molecular Docking Process
Molecular docking involves several key steps, which are outlined below:
1.?????? Preparation of Ligand and Receptor Structures: In most cases, the accurate three-dimensional structures of ligands and receptors are achieved through experimental methods like X-ray crystallography and NMR spectroscopy. If there is no experimental data available, molecular structures can be created using computational methods [12].
2. Grid Generation and Scoring Function: The binding site of the receptor, i.e., the site where ligands are supposed to bind, is decided either by manual or automatic methods. A grid is built around this site to check out different ligand positions and orientations. To evaluate the interaction between the ligand and receptor, a scoring function is employed. This function calculates the separation energy, which is the binding energy of the ligand-receptor complex [13]. The lesser the energy values, the tighter the binding and the more stable the interactions will be [14].
3. Search and Docking: The ligand is inserted into the receptor’s binding site and then it is systematically rotated and moved to examine different conformations. During this process, the scoring function is utilized to find the energy of the ligand for each position and orientation. Different search algorithms like genetic algorithms or Monte Carlo methods can be employed to better position the ligand thus finding the most stable and favorable binding conformation [15].
4. Scoring and Ranking: The ranking of these conformations is done based on the binding energy of ligands after their multiple conformations have been generated. Conformations having the lowest binding energy are regarded as the most stable ones and thus have the highest binding affinity. The drug candidates which are selected for further study and development are the conformations that have been top-ranked [16].
5. Analysis and Interpretation: At first, the emphasis is on the ligand conformations that got the highest ranks, then the researchers attempt to figure out how the receptor binds with the ligand. The ultimate goal of the research is to find hydrogen bonds, van der Waals forces, and electrostatic interactions. Moreover, the visualization tools assist in seeing binding orientations and interactions thus understanding molecular mechanisms and facilitating further ligand optimization [18].
6. Validation and Further Experiments: The binding poses of ligand-receptor from docking are confirmed by experimental methods, for example, X-ray crystallography or binding assays. If docking predictions are confirmed, research on the improvement of the ligand's binding affinity and selectivity through structure-based drug design and other optimization studies may be consequently pursued [19].
Among the serious problems of molecular docking, the fusion of quantum computing (QC) provides a range of potential solutions. One of the most significant limitations is the inability to recognize protein flexibility [20]. In general, a docking method assumes the rigid structures of the ligand and the receptor, a protein with an unchanged conformation, but proteins are still dynamic and have multiple conformations. The binding interactions can be changed significantly by these conformational changes [21]. The use of quantum computing makes it possible not only to consider multiple conformations but also their energetic contributions thus resulting in a more accurate representation of binding events and hence protein flexibility being modeled more precisely [22].
Conventional docking is also restricted by the simplification of solvation effects treatment. The solvent is the main player in molecular interactions and yet, in docking methods, the solvent is presented only as a simplified model, which is unable to capture the complexity of the solvent behavior. The classical preparations usually treat ligands and targets as static or semi-flexible, thereby ignoring solvation and entropic contributions, which lowers the prediction's accuracy [23]. Quantum computing would allow for the more realistic simulation of solvation effects, thus providing better insight into the stability and energetics of ligand-protein complexes [24].
Besides molecular docking is a computational method that heavily relies on the use of the computer, especially for large and complex biomolecular systems [25]. By virtue of their inherent parallelism [26], [27], and [28], and efficiency in quantum chemistry calculations, quantum computers can perform these calculations at a very high speed, thus docking studies take less time. Besides, quantum mechanics-based methods are more accurate than classical force fields; however, due to the high computational demands, these methods have been traditionally limited to small systems [29]. QC broadens the scope of quantum mechanical calculations to larger biologically relevant molecules thus leading to higher accuracy of binding affinity predictions [30].
Furthermore, quantum computing has the potential to radically change the chemical space exploration by more efficiently sampling a wider range of compounds [31]. Thus, the number of potential novel therapeutic molecules that classical methods would have overlooked is significantly reduced. The use of quantum computing in molecular docking is a way out of the problems raised by the classical methods which, in turn, means a tremendous potential for the progression of drug discovery and ??????development.
Table 1. Advantages of Quantum Simulations
|
Advantages of Quantum Simulation |
Description |
|
Higher Accuracy in Molecular Modeling |
Essentially the change is from a classical quantum picture of electrons and atoms to a quantum one which is direct. |
|
Faster Drug Candidate Screening |
In effect this technology is the best candidate to shorten the pharmaceutical research and development cycle by quickly filtering out most compounds that are not effective. |
|
Better Protein Folding Predictions |
Correctly representing a protein's energy landscape leads to the accurate prediction of protein folding is what has been achieved. |
|
Improved Drug-Target Binding Predictions |
If quantum forces (hydrogen bonds, van der Waals) are modeled accurately, then this will lead to more precise predictions of binding affinities. |
|
Rational Drug Design |
This system can be the first to enable the design of molecules with the desired characteristics such as solubility, chemical reactivity, and low toxicity. |
|
Reduced Costs and Development Time |
By removing the necessity for the process of trial and error, the drug development pipeline is accelerated resulting in reduced costs and time. |
|
Personalized Medicine Potential |
As a consequence, there will be the possibility of future medicine to harness the power of variations in individual genomes to develop tailored therapeutic ??????strategies. |
QUANTUM ALGORITHMS FOR MOLECULAR DYNAMICS SIMULATIONS
Molecular?????? dynamics (MD) simulations are computational models that depict molecular movements and interactions, thus enabling scientists to understand how new drugs bind to their biological targets. Nevertheless, conventional MD techniques are generally restricted in dealing with highly complex systems and might not be sufficiently accurate for making precise predictions.
A possible solution for such issues might be the use of quantum algorithms. These algorithms, which comply with quantum mechanical rules, can significantly speed up and make more accurate the computational part of MD simulations. Consequently, the time to market for new drugs would be shortened, treatment regimens would gain in effectiveness, and the expenses of the development process would be lowered.
Simply put, quantum algorithms for MD simulations are a way of using the incredible power of quantum computers to simulate molecules from the most fundamental level. Hence, they can be ultrafast and ultrahigh inaccuracy as they implicitly treat the quantum nature of molecular interactions [32] Fig ??????3.

Fig 3. Quantum Algorithms for Molecular Dynamics Simulations
1.?????? Wavefunction?????? Simulations: In quantum mechanics, the wavefunction is the model that defines probability amplitudes of different quantum states for both molecules and quantum systems. To figure out molecular properties and behavior one must first properly model such wavefunctions. It is a job that quantum computers perform in a fraction of the time by conceptually simulating the time evolution of wavefunctions. They can, therefore, interactively provide the whole energies and the dynamics of molecules. The consequence of this is that while classical computers are incapable of simulating quantum effects, wavefunction simulations are much more accurate and yield a more detailed understanding of molecular systems which in turn lowers the time required for trial-and-error processes in drug discovery and material design [33].
2. Quantum Phase Estimation: Before the understanding of molecular dynamics and chemical reactions can unfold, it is very important to have knowledge of the energy levels of a molecule. Quantum Phase Estimation (QPE) is one of the quantum algorithms that facilitate the finding of the eigenvalues of a quantum system; these eigenvalues are the energy levels of molecules. By producing these energy states with such high precision QPE in fact empowers the very exact modeling of molecular behavior, reaction pathways, and binding interactions. What is more, it extends the scope of drug discovery where scientists become able to simulate molecular interactions and even chemical changes of organisms that are going to take place with a level of accuracy far beyond that attainable by classical methods [34].
3. Quantum Walks and Quantum Monte Carlo Methods: Quantum Walks and Quantum Monte Carlo (QMC) are on the list of most outstanding quantum techniques that contribute to accomplish the understanding of molecular dynamics and thermodynamic properties of the system. The techniques in question in principle depict the movement, interaction, and changing structure of molecules at the energy minimum of the system as a whole. Because of the methods that they implement, they can simulate the toughest situations and in doing so depict the molecular stability, the reaction pathways, as well as the energetics in the more profound way. It is of great importance to drug discovery, where the understanding of the dynamics of drug-target interaction, in turn, leads to the production of effective and stable compounds, the core is [35].
4. Excited State Calculations: Through the use of quantum computers, scientists can tell the excited-state properties of molecules in such a way that they are almost perfect which lead to the uncovering of electronic transitions, energy transfer, and photochemical reactions. Quantum computing has enabled researchers to go far beyond the traditional limits in the comprehension of molecular behavior by precisely modeling the excited states and thus making predictions of the reactions of the molecules under various conditions become very simple. Such info is very crucial in pharmaceutical and material science fields where the change of excited states can bring up instability and reactivity issues or influence on molecular binding to biological targets ??????[36].
QML FOR VIRTUAL SCREENING
Virtual?????? screening is an in-silico method that makes use of computers to locate drugs from extensive chemical libraries. It goes through the molecular structures of a very large number of compounds that can even be in millions and chooses those molecules that have a high probability of binding to a target associated with a particular disease. Consequently, virtual screening alone is a time and cost-saving strategy in drug discovery.
Meanwhile, typical virtual screening techniques have some limitations. The prediction accuracy in simplified models is low and thus, a large number of compounds selected turn out to be inactive. These methods also demand a high level of computational power and the false positives that they produce need to be checked by other methods. Furthermore, the problems of discovering an enormous chemical space and considering complicated biological interactions remain.
On a theoretical basis, quantum simulations can eliminate all these issues. With the help of quantum computing, the researchers can perform virtual screening that is not only more precise but also quicker. Moreover, the use of quantum computing together with machine learning, known as quantum machine learning (QML), helps in the understanding of complicated molecular interactions and the identification of patterns [39]. QML exploits the benefits of quantum computing and machine learning to solve problems that are extremely difficult or even impossible for classical approaches, therefore, it can be a huge time saver in drug discovery [40].
The technologies based on quantum-enhanced computing that can help in virtual screening by making it more efficient and accurate are depicted in Fig. ??????4:
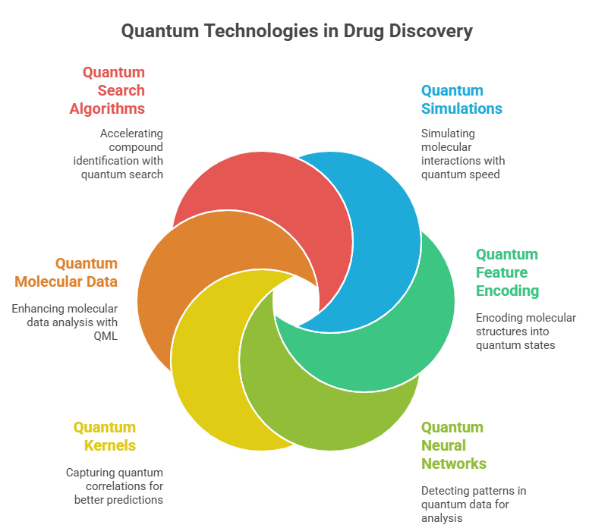
Fig 4. Quantum Technologies in Drug Discovery
1.?????? Quantum Simulations:
One of the most talked about applications of quantum computing is quantum simulation of complex systems can be done much faster and more efficiently by quantum computers than classical ones. In drug-discovery, precisely simulating molecular interactions is a must to figure out the complicated biochemical pathways. As a result, quantum simulations allow scientists to explore such interactions in great depth, to the extent that certain phenomena cannot be virtually demonstrated at all using classical approaches [41].
2. Quantum Feature Encoding:
The main idea behind the fast and effective analysis of drug candidates is the efficient representation of their molecular structures. In this regard, quantum machine learning (QML) can convert different molecular structures and their characteristics into quantum states directly, thus making data storage extremely compact and efficient which, in turn, boosts the virtual screening process [42].
3. Quantum Neural Networks:
By using quantum neural networks (QNNs), it is possible to uncover and learn the patterns of quantum data that might hard to be recognized by classical methods. These networks, which are realized by quantum circuits, not only store quantum information but they also manipulate it via quantum operations paving the way for more precise drug discovery through complex molecular interactions analysis and prediction [43].
4. Quantum Kernels:
Quantum kernels represent the quantum version of classical kernels employed in Support Vector Machines (SVMs). With this, they may include quantum features in the data that were ignored before leading to enhanced performance of machine learning techniques in drug discovery by precision predicting molecular interactions and new drugs.
5. Quantum Molecular Data:
Knowing structures and properties of complicated molecules is a process that usually involves methods like Nuclear Magnetic Resonance (NMR), X-ray crystallography in addition to other experimental techniques. Quantum machine learning (QML) can not only revolutionize a part of that but improve the speed and accuracy of those methods in drug discovery to an extent they will no longer be considered experimental.
6. Quantum Search Algorithms:
One category of quantum algorithms which comprises Grover’s algorithm is capable of looking through databases of potential compounds quickly and efficiently. In addition, by using quantum computing, these algorithms can perform the same tasks in much less time, thereby radically speeding up the initial selection process of the most promising drug candidates, thus making the virtual screening process faster and more effective [44].
Quantum machine learning (QML) techniques are likely to bring about cost reduction and time-saving in drug discovery. An example is a 2019 report by McKinsey & Company that examined the potential advantages of integrating quantum computing and machine learning into pharmaceutical research and development. The report studied different scenarios and projected that the company could achieve massive time and cost savings if it adopted these cutting-edge technologies in drug discovery processes ??????[45].
CONCLUSION
Quantum?????? simulations and quantum-enhanced computational techniques signify a fundamental change in drug discovery, as they offer ways to overcome the main limitations of traditional methods. In fact, these methods by accurately simulating molecular interactions and taking into account quantum effects improve the prediction of drug-target binding, optimize candidate molecules, and lower the number of experimental trials needed. Quantum computing makes molecular docking more accurate by taking into account protein flexibility, solvation effects, and complex energetic interactions, while quantum machine learning can be used to perform virtual screening in a much more efficient and accurate way over a large chemical space. In addition, quantum molecular dynamics, wavefunction simulations, quantum phase estimation, quantum walks, quantum Monte Carlo methods, and excited-state calculations are just some of the few examples of the unprecedented insights into molecular behavior, reaction pathways, and thermodynamic and electronic properties that can be gleaned. Together, these breakthroughs provide the means not only to speed up the identification and optimization of therapeutics but also to reduce the development costs and widen the drug discovery scope. The ongoing development of quantum technologies and their eventual adoption in pharmaceutical research is anticipated to revolutionize the efficiency, accuracy, and creativity potential of drug development by, among other things, enabling personalized medicine and the design of next-generation ??????therapeutics.
REFERENCES




Pocham Ravikiran*, Kethavath Mangulal, Mamidala Tejasree, Donthula Srilatha, Quantum Pathways to Innovation: Redefining the Landscape of Drug Discovery, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 39-53 https://doi.org/10.5281/zenodo.17775311
 10.5281/zenodo.17775311
10.5281/zenodo.17775311
