K. V. N. Naik S. P. Sanstha’s, Institute of Pharmaceutical Education and Research, Canada Corner, Nashik, 422002, Maharashtra, India.
The design of modern drugs has undergone rapid changes due to the development and improvement in computer science, molecular biology, and various analytical techniques. Most of the classical trial-and-error approaches are now supported by rational, targeted methods that enhance efficiency, specificity, and safety. This review describes current strategies in drug development, including new techniques for structure-based drug design, ligand-based drug design, computational drug design, fragment-based approaches, and artificial intelligence-based drug discovery.Integration of these approaches speeds the identification and optimization of new therapeutics by reducing cost and development time. This book addresses different aspects of the role of AI in drug development, including developing ways to use AI to deliver medicines. The process of pharmaceutical research and development has become more convenient since the introduction of many computer tools and software. Computer programs for online structural screening, ligand-based design, and lead optimization are often applied in drug development and discovery
The scientific approach to drug design focuses on discovering and developing new therapeutic compounds by understanding biological targets on a molecular scale. It generates molecules that can selectively engage with proteins or pathways associated with diseases by integrating ideas from chemistry, biology, and pharmacology. The objective of modern drug development is to formulate highly specific, safer, and more efficient therapies. Due to advancements such as structural biology, genomics, and computer-aided drug design (CADD), researchers can now develop medications more accurately and swiftly. This presentation explores the fundamental concepts, methods, and strategies employed in the process of drug discovery and design. Drug design is a collaborative approach focused on the identification of novel chemical compounds with target therapeutic effects and satisfactory safety standards[12]. Traditionally, drug discovery relied heavily on serendipity and the screening of natural products. However, escalating research costs, high attrition rates, and complex disease mechanisms necessitated more systematic approaches[13][14]. Modern drug development draws on chemistry, biology, bioinformatics, pharmacology, and computational modeling[15][16]. Advances in genomics and proteomics have revealed thousands of new drug targets, which have enabled precise molecular targeting. Meanwhile, computational methodologies have transformed the process for identifying, optimizing, and validating lead compounds before their experimental testing[17].
The research and development process in drug discovery is quite time-consuming and must consider the cost at every stage[18]. Traditionally, the ways in which agencies could test a particular chemical's effects on specific diseases were by increasing animal tests and adapting animal-based toxicity tests; these methods are very costly, labour-intensive, and require large numbers of animals. Most animal experiments are highly expensive and do not meet the set ethical criteria for research using animals.
History Of Drug Design
Ancient Beginnings
For a long time, even before written history, people utilized plants, minerals, and animal products to treat their ailments. This was frequently linked to convictions.
Paracelsus, who existed in the century, proposed innovative concepts regarding healing using plant extracts, minerals, and animal substances. He believed that the dosage of substances like metal salts, including mercury, can influence their effectiveness in treating ailments such as syphilis. Paracelsus focused on healing through the use of plant extracts, minerals, and animal products.
19th Century Developments
The identification of morphine from opium in 1806 by Friedrich Sertürner was significant. This was the moment when someone created a pure drug and experimented with it on animals. From 1820 to 1850, individuals could produce highly pure drugs such as quinine and atropine. This indicated that physicians could utilize medications rather than combinations of many substances. Morphine and these additional drugs were now distinct compounds, marking a shift from the earlier methods involving opium and various mixtures.
20th Century Achievements
Paul Ehrlich's salvarsan, developed in 1909, specifically targeted the disease known as syphilis. This constituted an agreement. Paul Ehrlich's salvarsan was the chemotherapy that effectively treated syphilis. Penicillin was found by Fleming in 1928. Following that, Florey and Chain focused on Penicillin. Improved it. They didn't limit themselves to a single city; they also altered our perception of antibiotics as a whole.
The design of medicines evolved with Paul Ehrlich's salvarsan. For instance, individuals created variations of ancient remedies such as aspirin, which resembles salicylic acid.
Modern Era
Since the late 20th century, structure-based design has been facilitated by X-ray crystallography, molecular modeling, and genomics. Methods such as de novo and fragment-based design are now producing new drugs aimed at specific protein targets.
The General Stages In Modern-Day Drug Discovery And Design
In the early nineteenth century, the discovery and design of drugs were left to individuals and relied more on chance than on systematic research. Over the last hundred years, a phenomenal expansion in our general scientific knowledge suggests that modern drug discovery requires intensive collaboration, with members being specialists in various fields such as medicine, biochemistry, chemistry, molecular modeling, pharmaceutics, pharmacology, microbiology, toxicology, physiology, and pathology[19]. The process is now more systematic, but a positive outcome still depends on some aspects of chance. The modern approach to drug discovery/design is goal-dependent. The scheme below (Figure 1) represents the general steps of the process involved in drug discovery and design
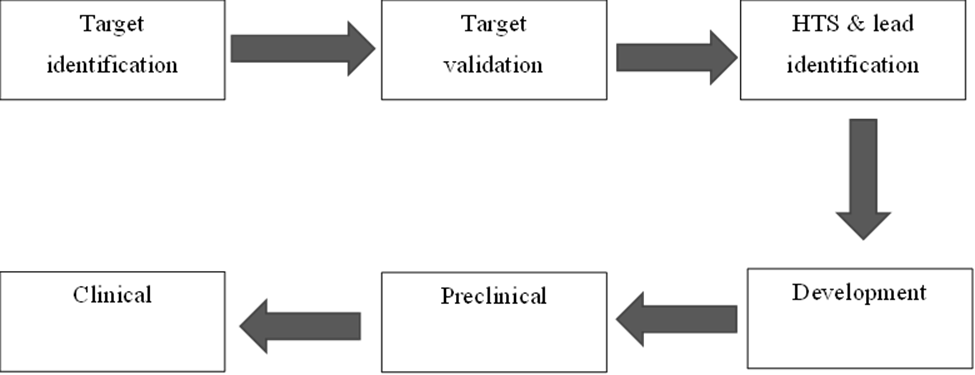
Fig.1. General stages in drug discovery and design
There Are Two Main Types Of Drug Design:
Rational Drug Design (RDD)
In the biopharmaceutical industry, rational drug design is a technique used to discover and develop new pharmaceutical compounds [1] [12]. RDD uses a variety of computational methods to discover new compounds, develop compounds for safety, efficacy and selectivity, and develop candidate compounds for clinical trials. Depending on how much information is available about the drug target and potential drug candidates, these methods can be divided into several categories, such as structure-based drug design, ligand-based drug design, de novo design, and homology modeling [12].
Structure-Based Formula Design (SBDD)
The three-dimensional arrangement of biological targets, especially proteins, is important for structure-based drug design [27] [28]. X-ray crystallography, NMR spectroscopy and cryo-electron microscopy are used to obtain structural data.[22]. One of the first approaches used in drug development was structure-oriented drug design [21]. Drug targets are usually important molecules involved in a particular metabolic or cellular signaling pathway known or thought to be associated with a particular disease. Proteins and enzymes in these pathways are the main targets for drugs [22]. The structure and function of proteins and enzymes associated with diseases are blocked, regenerated or otherwise altered by pharmaceutical compounds.
SBDD helps to create new drug compounds using the well-established 3D geometric shape or structure of the protein [23] [24]. This approach provides strong specificity and efficacy while enabling logical scaling of drug-target interactions [25].
Drug Discovery and Design Workflow:
The modern drug development process generally follows these methods [26]:
Main technologies for SBDD
Advantages:
limitations
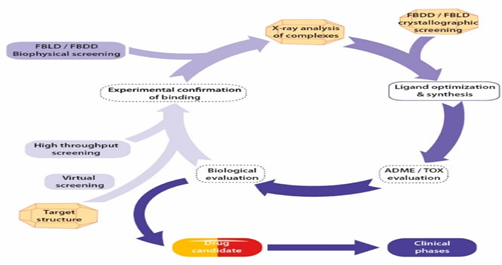
Fig.2. Integrated process of structure-based drug design(SBDD)
Ligand-Based Drug Design (LBDD)
Ligand-based drug design is employed while the target shape is unknown, yet active ligands are available[31]. This approach will become valuable when three-dimensional structural facts of the protein is unavailable but lively compounds are acknowledged[1].
Major Approaches
Advantages
Limitations
De Novo Drug Design
Drug development is an expensive process prone to error: only 1 in 5,000 candidates reach the market after preclinical and human testing. Innovative chemical devices are critical to maintaining the pipeline, but chemists struggle to explore the vast chemical space through high-throughput screening.
Computer-aided drug design (CADD) tools such as docking, QSAR and pharmacophore modeling accelerate discovery using structural data and computing power. Nevertheless, complex biology requires advanced methods.
De novo drug design (DNDD) generates new molecules from scratch using development algorithms, enabling comprehensive chemical exploration, IP creation and efficient candidate development. Main challenge: synthetic access.
This paper reviews the progress of DNDD—from traditional structure/ligand-based evolutionary methods (with limitations such as solubility or toxicity) to machine learning such as deep reinforcement learning—and outlines future integration with toxicogenomics and vaccines
De Novo Drug Design Methodology
De novo drug layout is a technique used to create new chemical materials primarily based entirely on information approximately a organic target, inclusive of a receptor, or on statistics about known energetic molecules that bind correctly to the goal. The important parts of this method involve developing a version of the receptor’s energetic website online or the key features of a ligand that allow it to bind, generating capability molecules, and assessing these generated molecules for his or her effectiveness. There are number one processes to de novo drug design: shape-based and ligand-based. Structure-based strategies depend upon three-dimensional fashions of the receptor, which can be received using strategies like X-ray crystallography, NMR, or electron microscopy. If the receptor’s structure is not known, homology odelling can be used to expect a suitable structure, even though the accuracy of this model depends on the first-class of the reference structure and the way similar the sequences are. Ligand-based totally strategies are regularly used when there is no structural data to be had for the target, however there are recognized molecules that bind to it efficaciously.
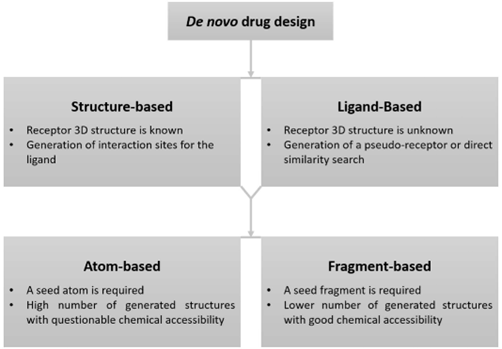
Fig.3. Schematic representation of the de novo drug-design methodology.
Fragment-Based Drug Design (FBDD)
Fragment-based drug design entails the screening of small, low-molecular-weight fragments that have weak binding interactions with targets[1][24]. This approach has gained significant attention in modern drug discovery due to its efficiency and broad coverage of chemical space.
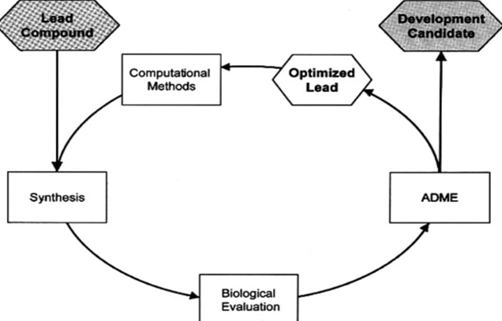
Fig.4. Iterative optimization pathway in fragment-based drug design (FBDD).
Advantages of FBDD
Obstacles:
Requires delicate detection techniques Fragment linking and optimization can be complicated. Artificial intelligence and machine learning in pharmaceutical development
Artificial intelligence has become a revolutionary influence in drug discovery.
Applications:
Benefits:
Constraints:
Computer-Aided Drug Design (CADD)
CADD integrates SBDD and LBDD through the use of computational tools[8][14]. This complete approach combines shape-based totally and ligand-primarily based strategies to decorate the drug discovery manner.
Essential Elements of CADD
Advantages
Molecular Docking
Predicting ligand-protein interactions, optimizing lead compounds, and virtually screening compound libraries are just a few the many application of molecular docking in drug discovery. Through scoring function and binding pose predictions, it aids in target exploration, finding new hits, repurposing existing drugs, and assessing polypharmacology or side effects [31][32]
Docking method in drug discovery
Jain (2017)[32] talked about using pc-aided strategies in drug discovery. It is a technique for predicting the place of tiny molecules or ligands within their target protein’s energetic place (receptor). The basic desires of molecular docking strategies are a prediction of ligand orientation concerning the receptor, power via which ligand and receptor bind, and virtual screening, all of that are interrelated. It provides a compound with a excessive hit price, and the probabilities of failure inside the final degree are very low. We can get understanding about drug interaction relationships. This approach can do away with the drug having low efficacy and poor ADMET belongings.
Mendy and Hemalatha (2022)[34] Using an in silico approach, this research focused on bioactive chemicals that can target GFR. Growth factor receptors (GFR) are proteins that are activated by interacting with their respective ligands (proliferation factors) and play important roles in tumor cell growth, angiogenesis, metastasis, cell survival, cell migration, cell death, differentiation, chemoresistance, neovascularization and organogenesis. Binding energy determines the binding affinity between receptors and ligands; The lower the energy, the stronger the bonding ratio. The binding energy (kcal/mol), the inhibition constant (M/nM) and the number of hydrogen bonds and amino acids involved in hydrogen bonding were all recorded after docking. This indicates that bioactive substances can bind to GFR, inhibit growth factors and, as a result, prevent cancer cell proliferation.
Kitchin et al (2004)[33] discussed scoring and docking methods in drug discovery. The docking method predicts the ligand configuration and orientation in a target binding site. From this method, we have two main conclusions: firstly, we know the structure of the model, and secondly, we know the activity of the drug molecule. When comparing docking approaches, it is important to consider how the ligand and protein are represented. The receptor can be represented in three ways: atom, surface and grid are used interchangeably. Virtual screening approaches, such as ligand-based virtual screening, allow de novo identification of active compounds without bias towards previous leads or hits. Although docking and scoring are based on a variety of heuristics, their use in lead optimization, usually in combination with other computational methods, already goes beyond more traditional approaches to structure-based design.
QSAR For Drug Discovery
Factor (2010)[35] discussed the role of QSAR methodology in drug discovery. Complex diseases, including single DNA variations (SNPs) in DNA, post-translational polypeptide changes, as well as climate-related factors play a significant role in disability and mortality on a global scale. Drugs, peptides, nucleotides, metabolic activity, diseases, and society are all examples of real dynamical structures that can be described statistically and compared based on the properties of the relationships between their component parts. Quantitative protein (or protein)-disease relationships (QPDR) and QSAR are used to predict medicinal effects and diseases, respectively. In 3 domains, cardiology and oncology, the study investigated the most relevant QSAR results in drug planning and application for complex diseases. Classification of molecules based on molecular properties and complex network/graph theory is used to generate mathematical models in a rapid QSAR approach as a tool for new drug formulation against serious disorders.
Winkler (2002)[36] mentioned the importance of QSAR in biomolecular discovery. By the QSAR approach, we can find out the molecule's 3-dimensional form. QSAR technique entails acquiring a descriptor from molecular structure, identifying the high-quality descriptor, mapping molecular form, and finalizing the excellent descriptor. Various techniques are available to generate descriptors. Selecting a descriptor is one of the maximum vital steps, but the usage of bad descriptors outcomes in various troubles in discovery. For mapping, the majority are regression strategies, the earliest of it truly is a couple of linear regression. And ultimately, it is vital to recognize how predictive the finalized structure is. Improved based totally totally on chemical entities, a better information of which molecular homes are most vital for a sure characteristic being simulated, and improved utilization of genetic and synthetic intelligence technology will increase QSAR's utility even better than it's miles now.
Rudrapal and Chetia (2020)[10] A quantitative structure-activity approach can be developed to generate quantitative correlations linking a structure to its biological activity (QSAR). The main principle of QSAR is to modify some compounds to improve their activity and create new compounds. Creating data sets, optimizing the structure, calculating and selecting molecular descriptors, and ultimately, model assessment and evaluation are all processes in QSAR analysis. Today, two types of QSAR are used 2D and 3D QSAR. A QSAR model must follow clear algorithmic methods and satisfy a number of tests, including model suitability, robustness and predictability. It serves as a tool to study the mechanisms of biological reactions using extraction. Especially in descriptions there are patterns. It is associated with the observed biological activity.
Zhang et al. (2006)[37] modeling strategy that was developed using the principle of latent training. The least squares statistical method and the bioactivity of substances in the test data set that are most chemically similar to the test compound are used to predict the effect of the test substance. An innovative Autonomous Lazy Leaning Quantification Structure-Activity Association (ALL-QSAR) modeling technique is built on the basis of lazy learning theory.
Quantitative structure activity relationship (QSAR)
In computational drug design, QSAR is an important chemometric tool[38]. QSAR research closely links the structural properties and the physical and chemical properties of structured compounds to their therapeutic activity. It tries to find a link connecting a series of experimental activity and molecular descriptors of compounds to define structural information. The response of biologically active chemicals to their structural, physical and chemical properties is represented by QSAR/QSPR models, which are mathematical equations.
Methods for quantitative structure-activity relationships have changed rapidly over the past decade. The main objective of this review is to summarize the status of QSAR with an emphasis on illustrating the utility and limitations of QSAR technology with examples of representative applications. Since it is impossible to cover such a broad subject in depth in a single communication, a robust bibliography is provided for the interested reader. We will first review 2D QSAR with a discussion of the availability and appropriate selection of molecular descriptors. We will then go on to describe 3D QSAR and the key issues associated with this technology, and then compare the relative suitability of 2D and 3D QSAR for different applications. Given recent technological advances in biological research for the rapid identification of drug targets, we cite several examples where QSAR approaches are used with improved knowledge of the structure and function of the target receptor. The review will conclude with a discussion of statistical validation of QSAR models, a topic that has received little attention in recent years despite its critical importance. In this regard, it is important to note that the utility of a QSAR model, regardless of the inherent sophistication of the methods used and the time taken to develop it, is only as good as the quality of the modeled data.
Rule:
QSAR variations
Applications
Advantages
Limitations
Pharmacophore Modeling
Pharmacophore modeling is the cornerstone of ligand-based fully computer-aided drug layout (CADD), which abstracts the spatial arrangement of molecular skills required in combination with hydrogen bond donors/acceptors, hydrophobic centroids, aromatic rings, and effective/terrible ionization organizations for a ligand to exert biological activity on its target.[45]
Modeling process
Ligand-based totally fashions are derived from aligning multiple energetic compounds to pick out not unusual pharmacophoric factors, regularly using tools consisting of LigandScout, Step or MOE with tolerance radii for bendy matching. Structure-based totally versions extract features at once from protein-ligand X-ray/NMR complexes, capturing pocket-specific interactions consisting of pi stacking or salt bridges. Validation uses metrics consisting of ROC-AUC on check sets, ensuring predictive electricity prior to database screening.
Pharmacophore Features
Categories
Applications
Advantages
Limitations
Pharmacokinetics and ADMET Considerations
Current drug improvement prioritizes early assessment of pharmacokinetic homes [27] [28]:
Early ADMET evaluation reduces the likelihood of errors at later tiers [32]. ADMET prediction using computational gear has grow to be an fundamental a part of lead optimization[33]. Obstacles and future potentialities Despite technological advances, issues persist:
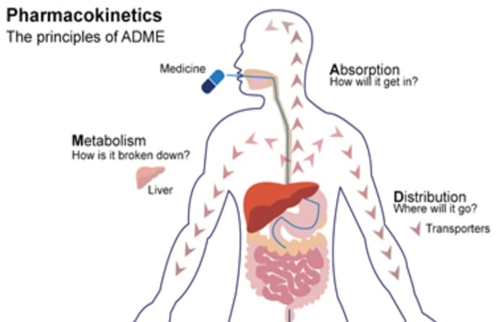
Fig.5. Role of ADMET in drug disposition and pharmacokinetics
The Impact of Natural Products on Modern Drug Discovery
Nature has, over time, created a very wide range of secondary metabolites[29]. Based on empirical evidence and traditional beliefs, natural product extracts were the first, and for a long time, the only medicines available to humans[11][30]. Although crude extracts still constitute the primary healthcare option for the majority of the global population, they are to a large extent replaced by active pharmaceutical ingredients in Western countries.
However, natural products remain valuable in drug discovery. Two main reasons can be propounded for the continued search of natural products as leads[30]:
Natural product-based approaches continue to contribute significantly to modern drug development, particularly in areas like anti-cancer and anti-inflammatory drug discovery[31][32].
Preclinical Testing
Preclinical studies and evaluation methodologies with and without animal testing are meant to minimize risks whenever a new active ingredient is being considered for administration in human beings as a medication[13][14]. They should be designed to allow an early and low-risk, simple and inexpensive transition from the preclinical to the clinical testing phases during medicinal product development.
Scientists perform both in vitro and in vivo tests[23]. In vitro experiments are done in the laboratory and usually involve test tubes and beakers ("vitro" means "glass" in Latin), while in vivo studies utilize living cell cultures and animals ("vivo" is Latin for "life"). Advanced model systems like zebrafish have become valuable tools in preclinical drug evaluation[24][25].
Clinical Testing
A clinical trial (or research study) is a scientific study conducted on human volunteers to answer specific health questions[26]. Well-designed medical trials are the safest and fastest way to find effective treatments and ways to improve health. In a clinical trial, researchers:
The National Institutes of Health (NIH) classifies trials into five different types: prevention trials, screening trials, diagnostic trials, treatment trials, and quality of life trials[26].
NDA and FDA Approval
The New Drug Application (NDA) in the United States is the vehicle through which the drug sponsors request the FDA to approve a new medication for marketing and sale[27]. This NDA is designed to contain adequate information for the FDA reviewers. The NDA will summarize all the information from preceding years of work and also includes the plans for the manufacturing and labeling of the new drug in question.
FDA specialists review all the documentation included in the NDA to judge whether or not it demonstrates that the drug is safe and effective enough to be approved[28]. This rigorous review process ensures that only medications meeting stringent safety and efficacy standards reach consumers.
Applications of drug design
Future Outlook and Challenges
Issues continue despite technological progress[26][27]:
The design of future drugs is anticipated to depend significantly on integrated AI systems, tailored medicine, and multi-target therapeutic strategies[19][31][33]. The promise is guaranteed by the ongoing advancements in pharmaceutical research, such as automated synthesis platforms, generative diffusion models, and advanced animal models like zebrafish [24][25].
CONCLUSION
The discovery of new drugs and the development of those drugs take a long time and are very costly[26]. After setting goals, the work starts with confirming targets and identifying drug candidates. Any novel drug is subjected to preclinical and clinical trials, and only after FDA approval can it be sold to consumers[27][28].
Machine learning has become a powerful instrument in drug discovery, making new treatment methods more feasible for different diseases[19]. The involvement of ML-based methods such as deep learning and neural networks has been instrumental in discovery activities, thus cutting down both the time and the amount of money committed in traditional drug development[8][14][29].
Additionally, computational tools and software have improved research and development (R&D) in pharma through online screening, structure-based design, and lead optimization[9][10]. The pharmaceutical sector is in perpetually changing state; thus, one may foresee that increased demand for ML applications will be the main driver that leads to concentration of the future of healthcare in ML-based drug discovery[31][33].
Among other ways in which AI and deep learning models have been improved over time is their use for drug discovery[19]. The models can now be used in target identification, prediction of drug-target interactions, and optimization of pharmacokinetic and toxicity profiles for drugs very quickly[14][29]. Moreover, phenotypic screening, omics-based target identification, and systems biology methodologies provide comprehensive understanding of disease mechanisms, thereby enabling the manufacture of more precise and personalized medications[25].
The continuous innovations in pharmaceutical research, including generative diffusion models, automated synthesis platforms, and sophisticated animal models like zebrafish, ensure that the potential for future drug discovery remains limitless[24][25][31].
REFERENCES



Jagruti Gosavi, Trupti Kadam, Avinash Darekar, An Overview of Modern Drug Design Approaches, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 4305-4320. https://doi.org/10.5281/zenodo.18110852
 10.5281/zenodo.18110852
10.5281/zenodo.18110852
