1-3Students, Ashokrao Mane Institute Of Pharmacy, Ambap, Kolhapur-416112
4Assistant Professor, Ashokrao Mane Institute Of Pharmacy, Ambap, Kolhapur-416112
5Professor, Ashokrao Mane Institute Of Pharmacy, Ambap , Kolhapur – 416112.
Preformulation studies are a crucial step in the pharmaceutical and drug development process. These studies provide a comprehensive understanding of the physicochemical properties of a drug candidate, guiding subsequent formulation and optimization efforts. This overview highlights the significance of preformulation studies in drug development, their key components, and their role in ensuring the success of pharmaceutical products. Preformulation studies encompass a range of investigations, including drug characterization, stability assessment, and formulation compatibility evaluation. Characterization involves determining the drug's physical and chemical properties, such as solubility, partition coefficient, and crystallinity. Stability studies assess the drug's degradation kinetics under various conditions, aiding in the establishment of appropriate storage conditions and shelf life predictions. Compatibility studies examine the drug's interaction with excipients, ensuring that the chosen formulation components do not adversely affect the drug's stability or efficacy. The outcomes of preformulation studies enable formulation scientists to design efficient drug delivery systems, select appropriate excipients, and optimize drug formulations. By addressing potential challenges and risks early in the development process, preformulation studies contribute to reducing development costs and time-to-market.
Drug:
A medication is a chemical compound designed for therapeutic purposes. As it is impossible for it to be ingested in its unaltered form, it is processed to appropriate administration formats to ensure secure and effective administration within the body [1].
Preformulation:
Pre-formulation studies are conducted with the aim of collecting comprehensive data, specifically pertaining to the various properties of active ingredients, excipients, and packaging materials used in therapeutic products [2]. At the initial phases of formulating a novel drug, preformulation typically involves a comprehensive examination of the active pharmacological component and other components employed in the composition. This characterization encompasses various aspects [3]. To aid in the advancement of a dependable and efficacious composition, the careful choice of excipients holds paramount importance [4]. The most common indications of degradation in an active pharmaceutical ingredient (API) include alterations in color, taste, odor, polymorphic structure, and crystallization, which are often associated with pharmaceutical incompatibility. These changes are typically the result of chemical interactions with excipients [5]. After identifying a pharmacologically active compound, a project team, comprising experts from various fields, is tasked with ensuring that the substance proceeds into the development phase in its most advantageous molecular state [6]. The selection of excipients must be conducted with precision to create a sturdy and efficient formulation for dosage forms [7, 8]. In the late 1950s and early 1960s, there was a notable shift in the focus of industrial pharmaceutical product development, which introduced the concept of preformulation. [9]. The preformulation study is an integral component of an interdisciplinary approach [10]. In facilitating creation of a robust product, this study incorporates initial drug degradation profiles. Following the completion of biological assessments for the medication and the decision to proceed with its development in clinical trials, the formal preformulation study commences. The guidance for preformulation studies is provided by the recommendations outlined in the IND, NDA, and ANDA guidelines issued by both the ICH and the US FDA [11]. Why do many people readily embrace various forms of medication without scrutinizing their safety? It's because of their trust in us and the formulations we create. As pharmaceutical scientists and formulation chemists, it is our responsibility to meet their trust by ensuring the guaranteeing the safety and effectiveness of these goods they use. For marketing clearance, the FDA has received the submission of the final product and demonstrates consistent chemical and physical attributes, it signifies a positive step forward [12].
Importance of Dosage Forms in Medication:
OBJECTIVES:
KEY ASPECTS IN PREFORMULATION RESEARCH STUDIES [14, 15, 16].

Bulk characterization-
At this stage, the solid forms of a potential drug are not yet fully characterized, leaving ample space for the discovery of new polymorphic structures.
Crystallinity and polymorphism:
Elements can exist in multiple distinct forms known as allotropes. Just as the crystal structures of two different compounds vary, various polymorphs of a single chemical substance typically possess distinctive characteristics. The occurrence of polymorphism is a widespread phenomenon in organic molecules, and numerous medications have the capacity to crystallize into diverse polymorphic structures [17]. In reality, it has been shown that variations in solubility across polymorphs are often less than a factor of two or less frequently, five [18, 19, 20]. In practice, it's common for metastable and more soluble forms to undergo a swift transformation into the thermodynamically more stable state. [21, 22]. The ongoing progression of this transition relies on the relative thermodynamic stability of the metastable states [23]. When water is employed as the solvent within a solvate, it's termed a hydrate. In pharmaceuticals and drug development, the utilization of solvates is generally discouraged due to potential toxicity associated with the existence of leftover organic solvents [24]. Hydrates may exhibit varying dissolution rates in comparison to their anhydrous counterparts. Typically, the dissolution of hydrates is slower, possibly due to the limited availability of pharmacological component sites engaging with water throughout the procedure [25, 26].
Type of Polymorphism-
In the case of polymorphs, one may be stable under particular variables of force and heat levels , while the other becomes stable under different pressure and temperature conditions, as exemplified by sulfur.
Among multiple polymorphs, only one remains stable at temperatures below the melting point, while all the others are found to be unstable, such as in the case of chloramphenicol palmitate with glyceryl stearate
The term 'pseudo' signifies 'false.' Solvates involve the integration of solvent molecules into the solid's crystal lattice. Such solvates can manifest in various crystal forms, referred to as pseudopolymorphs, and this phenomenon is termed pseudopolymorphism [27].
Hygroscopicity : -
Many chemicals and salts can be vulnerable to moisture or water vapor. When these substances come into contact with moisture, they can retain it through mechanisms such as surface adsorption, capillary condensation, chemical reactions, and, in extreme cases, deliquescence. Moisture represents a significant element that can influence constant nature of proposed medications and compositions. Interactions with water molecules with either a potential medication or additives can often lead to hydrolysis. In such instances, contact between the drug and excipients may result in ionization of one or both molecules, potentially causing a chemical reaction [28].
Fine Particle Characterization: -
Obtaining a representative dispersion necessitates careful sampling and meticulous preparation of the microscopic slide. In tandem with light microscopy, the utilization of stream counter devices such as the Coulter counter and HIAC counter are commonly employed to analyze the size distribution of substances. For larger samples with relatively large particles (typically around 100 microns), sieve techniques are the preferred method [29].
Flow properties:
A smooth flow of drug ingredient powder is essential for the effective formation of tablets. One of the key reasons for considering this characteristic in preformulation is its interconnectedness in conjunction with other psychological factors, such as hygroscopicity, particle size, and shape. The flow characteristics of solid pharmaceutical materials are commonly evaluated using parameters such as the Hausner ratio, Carr's index, and the angle of repose.
The Carr's compressibility index is expressed as:
Carr’s compressibility index = Tapped density-Bulk density Tapped density
Hausner’s ratio can be represented by:
Hausner’s ratio = Tapped densityBulk density
Table.1. Correlation between relative humidity, moisture uptake, and flowability
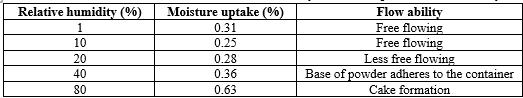
The correlation between Carr's index, Hausner's ratio, and flowability can be observed in Table.2. An alternative method for assessing flow characteristics is by using the angle of repose, which supplies insight into particle refusal to motion. It can be quantified using the formula:
Tan ? = 2h/D
Table 2. Correlation between Carr’s index, Hausner’s ration, and flowability

The angle of repose represents the maximum angle achievable between the pile's height and the horizontal plane [10].
Solubility analysis:
The primary goal of preformulation efforts is to establish a method for producing pharmaceutical solutions. For a drug to be therapeutically effective, it needs to be soluble in water. Before a medication can gain access to the bloodstream and elicit its medicinal action, initially exist in a dissolved state. In cases where substances are somewhat insoluble, incomplete absorption can occur. Preformulation involves the study of interactions between a drug and its solvent that can occur during drug administration. For instance, orally administered drugs should undergo solubility testing in a simulated stomach medium. Analyzing the solubility of a new medication is essential to lay the groundwork for future formulation work and understand how the drug will perform. Medications with an aqueous solubility below 1% (equivalent to 10 mg/ml) may encounter bioavailability challenges [30].
Ionization consant-pKa :
The measurement of pKa or ionization constant holds significant importance in the pre-development phase, particularly because a majority of drug candidates are weak acids or bases. The ability of acidic and alkaline compounds to dissolve is contingent upon the pH of the medium. Strong acids, like HCl, ionize at all pH levels, while the Ion formation from weak acidic compounds is pH-dependent.
For basic compounds:
pH=pKa+log ?((concentration of ionized species)/(concentration of unionized species))
For acidic compound:
pH=pKa+log ?((concentration of unionized species)/(concentration ofionized species ))
At the point where pKa equals the pH, there is typically a balance with approximately 50% dissociation (ionization) and 50% unionization of the substance [31].
Common ion effect (Ksp):
The common ion effect is a solvent interaction that is commonly encountered but often overlooked. The presence of a shared ion often decreases the solubility of a moderately soluble electrolyte. This phenomenon occurs as water molecules are 'crowded out' by competing ions through hydration, leading to a process known as 'salting out.' Consequently, weakly basic drugs administered exhibit lower solubility in an acidic solution [32].
Solubilization :
A common approach to enhance solubility encompasses the introduction of a co-solvent into the water-based solution. The use of suitable co-solvents like ethanol and glycerin often results in a substantial increase in relation to the solubility of substances that have limited dissolving non-electrolytes, sometimes by a vast difference [33]. Ethanol, sorbitol, glycerin, and PEG are among the most commonly employed and suitable co-solvents for formulating aqueous oral solutions. Dimethylacetamide is often used in parenteral products, but its use in oral solutions is limited due to its unpleasant odor and taste. It's worth noting that the co-solvent effects are typically considerably weaker for dissociated pharmaceutical compounds [34].
Dissolution:
The absorption of sparingly soluble medications are commonly more influenced by its dissolution rate rather than its saturation solubility. Hence, it is of paramount importance to experimentally determine the dissolution rate.
Intrinsic dissolution:
Breaking down the Noyes-Nernst equation provides a precise representation of a solid's rate of dissolution in its own solution. The parameter 'Mg dissolved (min^-1 cm^2)' is frequently employed to depict the intrinsic dissolution rate within a consistent volume of solvent.
Particulate dissolution:
This assessment will determine the drug's dissolution performance across different surfaces. It serves as a tool to explore the impacts of factors like particle size, surface area, and the blending of excipients on dissolution. In cases where a particle dimension has minimal contribution to the dissolution rate, alternative approaches, such as the inclusion of surfactants [32].
Partition coefficient :
The partition coefficient, denoted as log P, is a common measure used to characterize the lipophilicity of an organic molecule and is defined as the equilibrium of the distribution of the non-ionized compound concentration between the organic and aqueous phases
Log P =(unionized compund )org(unionized compund)org
Complex lipids within biological membranes are difficult to isolate in their purest form. Nevertheless, the relative lipid solubility of a pharmacological ingredient can be deduced by observing how it disperses between water and an immiscible organic solvent.
Methods offending Partition coefficient:
Stability Analysis:
Solution stability:
The degradation of a drug in its solution form typically occurs more rapidly compared to its dry form. It is crucial to ensure that the medication remains stable under the conditions of gastrointestinal (GI) fluid. A stability investigation with a focus on ph can be designed, employing various GI fluid simulators. In cases where the drug's solution stability is compromised, the formulator might consider choosing a less soluble salt form, provided that it does not negatively impact the drug's bioavailability [6].
Solid state stability:
The main purposes of this investigation include the exploration of optimal storage parameters for
drugs in their solid phase and the excipient characterization that exhibit effective compatibility in formulations. Both excipients and the drug itself contribute some free moisture to all solid dose formulations, with tablets typically requiring a substantial amount, often around 2% w/w, to achieve the necessary compression. The initial quantitative assessment of a new drug's chemical stability is conducted through the stability testing of pharmaceutical products. This process involves evaluating a specific formulation's ability to preserve its physical, chemical, microbiological, pharmacological, and toxicological attributes throughout its intended shelf life when stored in a specified container or closed system. [35].
Physical properties:
Crystalline Vs amorphous form:
In amorphous pharmaceuticals, molecules or atoms are randomly arranged within the chemical lattice. Common methods for generating typical amorphous forms include solid particle settling, quick cooling following the molten state, and freeze-drying. Among the highly remarkable aspects significant benefits of the amorphous form is its enhanced dissolving capacity, leading to an increased dissolution speed. Low water-soluble drugs often exhibit poor bioavailability and inconsistent therapeutic outcomes. In such cases, polymorphic forms can often address this issue.
Table 3. Correlation of solubility and BCS class and associated approach in
Soluble compounds are often characterized by their stability. It's worth noting that amorphous forms tend to have reduced stability compared to their crystalline counterparts, and they often transform into more stable forms when stored for extended periods. Nonetheless, the trade-off between risks and advantages still favors the use of amorphous materials, making them a preferred choice for product development. [36].
Particle size and size distribution:
The particle size distribution and forms of pharmacological compounds can influence various chemical and physical properties. In some instances, these effects extend to the biopharmaceutical behavior of solid drugs, in addition to their physical characteristics. For example, the particle size distributions of griseofulvin and phenacetin have a direct impact on their bioavailability. The size of the active ingredients and excipients can also affect the evenness of the tablet's composition [28].
Stability profile:
Water sorption often conforms to a Type-II isotherm, allowing for the selection of humidity levels both below and above the isotherm's threshold. For example, this may include conditions such as 30% and 75% relative humidity (RH) at a moderately elevated temperature of 40°C [37, 38]. The stability evaluation necessitates a specific quantity of the drug substance, which varies depending on the analysis techniques employed. As a standard, one can calculate the requirement to be 500 mg for each pull time, resulting in a total of 2.5 g for the study conducted at 40°C under two different relative humidities. The evaluation of light stability is conducted concurrently with dark stability testing. To assess stability under particular humidities, a testing program involving up to 48 hours of light exposure can be carried out using simulated sunlight. If the same relative humidities as those used in the dark stability research, specifically 30% and 75% RH, are applied, a total of 2.5 g of material is required, considering a single temperature and two exposure durations [39]. By employing tristimulus colorimetry, it becomes feasible to assess the surface color both before and after exposure to light [40].
Differential scanning calorimetry (DSC):
For over five decades, the utilization of Differential Scanning Calorimetry (DSC), a prevalent method in the field of thermal analysis, has been steadily growing as part of the assessment for compatibility for active pharmacochemical components. DSC holds an advantage in contrast to standard techniques as it demands lower sample quantities and shorter analysis times. Furthermore, it provides valuable indications of potential issues, allowing for the early exclusion of an excipient in the product design and creation procedure. When the use of the excipient is imperative, additional investigations can be conducted to understand the way it interacts with the API [41, 42].
Fourier-Transform Infrared Spectroscopy (FTIR):
A spectrophotometer is a tool used to measure the absorption spectrum of a substance. In comparison to traditional spectrophotometers, Fourier transform spectrophotometers deliver the more rapid IR spectrum measurement. The mid-IR region, spanning from 4000 to 666 cm^-1, encompasses the IR spectrum generated by the FTIR spectrometer. The presence of an absorption band in the mid-IR region (4000-400 cm^-1) can be employed to determine the existence of specific functional groups within a molecule, as many functional groups exhibit energies linked to alterations in vibrational energy states in this region (4000-400 cm^-1). FTIR spectra are capable of analyzing four distinct regions, each associated with different bond types. Single bonds (OH, CH, and NH) are observed at higher wavenumbers (2500–4000 cm^-1). In the intermediate wavenumber ranges between 2000 and 2500 cm^-1 and 1500 and 2000 cm^-1, triple bonds and double bonds become apparent, respectively. The identification of the entire molecule can be achieved in the low wavenumber range between 650 and 1500 cm^-1.
Advantages of FTIR:
Disadvantages of FTIR:
DRUG EXCIPIENT COMPATIBILITY STUDY BY USING FTIR AND DSC METHODS DSC METHOD:
The temperature and heat flow changes associated with material transitions can be determined by employing a thermodynamic method known as Differential Scanning Calorimetry (DSC). This method proves highly effective in assessing a medication's physical properties, its interactions with other substances, and the stability of the substances within the ultimate dose type. DSC is particularly valuable in the detection of possible incompatibilities, as it can reveal alterations in the curves, including the visual appeal, disappearance, or shift of heat-absorbing or heat-releasing peaks, along with deviations in heat flow values [47].
Experiment:
The DSC thermograms for modafinil and its physical mixtures were recorded using a Mettler Toledo AG DSC 821 machine in a nitrogen atmosphere with a flow speed of 50 mL/min. The observations were taken in the thermal range of 20°C to 220°C at a heating rate of 10°C/min. Additionally, non-isothermal studies were undertaken using different thermal speed within the same temperature range, specifically at 5, 10, 15, and 20°C/min. A mass of 4.0 mg (±0.1 mg) was weighed for each sample, and the DSC underwent calibration with indium as the reference material. The crucible used for these measurements was hermetically sealed.
RESULT AND DISCUSSION:
Figure given depicts the differential scanning calorimetry (DSC) curves for modafinil in isolation and modafinil combined with various excipients. Modafinil showcased a prominent melting peak at 170°C, followed by rapid degradation, suggesting its thermal stability before the onset of melting, with no other preceding thermal events observed. However, the DSC curves for the formulations containing excipients displayed noteworthy alterations in the melting endotherms and degradation exotherms. The melting point of modafinil was observed to shift to approximately 5°C lower in the presence of MgSt, indicating a potential interaction between the two components. It is important to note that such fluctuations in melting points could be attributed to variations in material purity during the mixing process, rather than necessarily indicating incompatibilities between the component [48].
FT-IR METHOD:
A simple and effective method for detecting changes in pharmaceutical-excipient combinations is Fourier-Transform Infrared Spectroscopy (FTIR). The presence of involvement among the active pharmaceutical ingredient (API) and the additives under investigation is typically noticed by observable changes in the FTIR spectra. These changes may manifest as the elimination of specific consumption peaks, a decline in the level of exertion of existing peaks, or the emergence of new peaks in the spectra. FTIR is a effective instrument for assessing compatibility and interactions between drug substances and excipients in pharmaceutical formulations [49, 50].
Experiment:
In the study, an FTIR spectrometer, specifically a Nicolet iS10 FTIR spectrometer, was employed to obtain infrared spectra within the range of 4000-400 cm??1;. The solid speciemens were positioned directly on a diamond attenuated total reflectance (ATR) accessory known as Smart iTR, without the need for crushing or mixing with KBr. Spectra were collected by performing 32 scans at a resolution of 4 cm??1;. For the compatibility testing, tobramycin and its physiological compositions including additives are enclosed in a glass package and subjected to different storage conditions: room temperature, 40 °C, and 75% relative humidity, and 40 °C and 75% relative humidity in which 100 ?L of purified water is added, over a period of four days. Prior to capturing the infrared spectra, the specimens were vacuum-dried at 40 °C [51]
RESULT AND DISCUSSION:
In the Fourier transform infrared spectroscopy (FTIR) spectrum of pure tobramycin, several characteristic bands were observed:
After subjecting tobramycin to different storage conditions (specifically, exposure at 40 °C and 75% relative humidity for 4 days, both with and without the addition of water), the FTIR spectra of tobramycin remained unchanged. This indicates that there were no observable modifications or interactions between tobramycin and the storage conditions during the four-day exposure period [52].
CONCLUSION
Preformulation studies are a critical foundation for the successful growth of pharmaceutical dose types. These studies serve as the initial stepping stones in the rational plan of stable, efficient, and methods for protected delivery of medications. By gaining a deep understanding of the physical and chemical properties of a drug candidate, scientists and formulators can make informed decisions about the choice of excipients, processing techniques, and drug forms. One crucial aspect of preformulation research is the exploration of polymorphism, which is common among organic molecules, especially in the context of pharmaceuticals. Polymorphs and other solid forms of drugs have distinct physicochemical properties that influence solubility, stability, and ultimately, bioavailability. Solubility is a primary concern in preformulation, as it directly impacts a drug's effectiveness. Researchers investigate various factors affecting solubility, including pH, temperature, and the use of co-solvents. Additionally, preformulation research examines other essential parameters like compatibility between active pharmaceutical ingredients and excipients, thermal analysis, and stability studies. All these elements collectively contribute to the growth of pharmaceutical dose types that are safe, efficient, and meet the needs of patients and healthcare providers.
ACKNOWLEDGEMENT:
I would like to express my special thanks of gratitude to our principal Sir, Ashokrao Mane Institute of Pharmacy, Ambap. I am really thankful to them to Mr. Sardar S. Shelake Sir for the patience and support in every Methodological aspect of this several other works. Thank you for your honest advices, your help in overcoming so many obstacles, and above all thank you for all your interest. Secondly I would also like to thanks my friends who helped me a lot in finalizing this project within the limited time frame. Last but not the least, my parents are also an important inspiration for me so with due regards, I express my gratitude to them also.
CONFLICT OF INTEREST STATEMENT:
The authors declare that they have no conflicts of interest regarding the publication of this review paper. All authors certify that they have no financial or personal relationships with other people or organizations that could inappropriately influence (bias) their work or this manuscript.
REFERENCES

Komal Bhusare , Prathamesh Regade , Nikita Patil , Sardar Shelake , Nilesh Chougule , Preformulation Studies An Overview , Int. J. of Pharm. Sci., 2024, Vol 2, Issue 7, 984-996. https://doi.org/10.5281/zenodo.12739436
 10.5281/zenodo.12739436
10.5281/zenodo.12739436
