MVP Samaj’s College of Pharmacy, Nashik – 422002, Maharashtra, India.
Poor aqueous solubility remains a major challenge in pharmaceutical drug development and is a leading cause of low bioavailability and formulation failure, particularly for drugs belonging to Biopharmaceutics Classification System (BCS) Class II and IV. Traditional solid-state approaches such as salt formation, polymorphism control, and amorphization often suffer from limitations related to stability, manufacturability, or regulatory constraints. Pharmaceutical co-crystals have therefore emerged as an effective and versatile solid-state modification strategy capable of modulating physicochemical and biopharmaceutical properties without altering the molecular structure of the active pharmaceutical ingredient. This review integrates fundamental crystal engineering principles with pharmaceutical applications, focusing on supramolecular synthon theory and the role of non-covalent intermolecular interactions such as hydrogen bonding, halogen bonding, ?–? interactions, and van der Waals forces. Various co-crystallization techniques, including mechanochemistry, hot-melt extrusion, supercritical fluid processing, microwave-assisted synthesis, and continuous manufacturing, are discussed alongside their pharmaceutical relevance. Applications related to solubility enhancement, bioavailability improvement, stability modulation, taste masking, and regulatory considerations are highlighted, positioning pharmaceutical co-crystals as a rational and scalable solid-state strategy for modern drug development
1.1 Background and Problem
The modern pharmaceutical industry faces alarmingly high attrition rates in drug discovery and development, with a substantial proportion of failures occurring during the transition from preclinical studies to clinical trials. One of the most critical underlying causes is poor aqueous solubility and low dissolution rate of Active Pharmaceutical Ingredients (APIs), which directly limit the in vivo bioavailability of otherwise promising drug candidates. It is estimated that approximately 40 % of currently marketed drugs and up to 90 % of compounds in development pipelines suffer from solubility-related challenges [1, 2]. The problem is particularly severe for Biopharmaceutics Classification System (BCS) Class II and IV drugs, whose absorption is dissolution-rate-limited [3]. These solubility issues hinder efficient gastrointestinal absorption, resulting in sub-therapeutic plasma concentrations and poor clinical outcomes [4].
Low solubility is not merely a formulation inconvenience; it is a multi-dimensional problem that affects the entire drug-development pipeline. Poor dissolution profiles increase variability in pharmacokinetic parameters, reduce dose proportionality, and necessitate higher doses—potentially leading to increased side effects and patient non-compliance. In addition, solubility limitations can delay regulatory approval, increase manufacturing costs, and jeopardize the commercial viability of otherwise effective drug molecules [5].
Traditionally, pharmaceutical development followed a linear and siloed approach, sometimes described as “throwing molecules over the wall.” Candidate molecules moved sequentially through discovery, pre-formulation, and formulation development with limited early integration of solid-state characterization. This practice often advanced compounds into clinical stages before thorough evaluation of their polymorphism, salt forms, hydrates/solvates, or co-crystal potential had been conducted [6].
Insufficient early-stage solid-state screening frequently leads to late discovery of critical manufacturability and stability issues such as:
Such issues result in costly delays, reformulation needs, and even project termination. This underscores the urgent need for integrating solid-state screening and optimization strategies early in drug development, enabling risk identification and mitigation before critical late-stage failures occur [7].
1.2 Pharmaceutical Solid-State Forms
Active Pharmaceutical Ingredients (APIs) may exist in multiple solid-state forms, each possessing distinct physicochemical properties that influence solubility, dissolution, bioavailability, stability, and manufacturability [8]. Control and optimization of these forms are vital, as crystal packing and intermolecular interactions directly affect a drug’s performance both in formulation and in vivo [9]. The principal solid-state categories—polymorphs, salts, hydrates/solvates, amorphous solids, and co-crystals—offer diverse options for tuning biopharmaceutical behaviour [10].
1.2.1 Polymorphs
Polymorphism is the ability of a compound to crystallize in two or more lattice arrangements while retaining identical chemical composition [11]. Distinct polymorphs may exhibit markedly different melting points, densities, and dissolution rates; metastable forms often dissolve faster but are less stable. From a regulatory standpoint, complete polymorphic characterization is mandatory, as unexpected transitions can alter therapeutic performance [11, 12]. The notorious ritonavir case, where a more stable yet less soluble polymorph emerged post-approval, exemplifies this risk [12].
1.2.2 Salts
Salt formation remains a primary route to enhance solubility and dissolution [13]. By pairing an ionizable API with an oppositely charged counter-ion, new crystalline lattices of superior aqueous solubility and compressibility can be obtained. However, inappropriate counter-ion selection may lead to hygroscopicity or incompatibility issues [13]. Typical marketed examples include amlodipine besylate, sertraline HCl, and metoprolol tartrate.
1.2.3 Hydrates and Solvates
Hydrates and solvates incorporate water or solvent molecules within the crystal lattice [14]. These inclusions modify melting point and dissolution behaviour: hydrates are often less soluble due to hydrogen-bond stabilization, whereas some solvates display improved wettability [14]. Loss or exchange of solvent during storage can trigger structural collapse; hence, controlling crystallization and humidity is essential.
1.2.4 Amorphous Solids
Amorphous forms lack long-range order, possessing higher free energy and, consequently, higher apparent solubility [15]. Because no lattice must be disrupted, dissolution is faster; yet their thermodynamic instability predisposes them to recrystallize on ageing or under humidity. Stabilization strategies employ polymers such as PVP or HPMC that inhibit molecular mobility [15].
1.2.5 Co-Crystals
Pharmaceutical co-crystals consist of an API and a neutral co-former linked by non-ionic, non-covalent interactions such as hydrogen bonding, halogen bonding, or π–π stacking [16]. They extend solubility-enhancement possibilities to non-ionizable drugs and can simultaneously improve mechanical and stability profiles [16, 17]. Co-crystals differ from salts by the absence of proton transfer and are now recognized by the U.S. FDA as distinct crystalline forms, supporting new intellectual-property claims [17].
Summary of Solid-State Forms
Selecting the optimal solid-state form depends on the targeted improvement in performance and the physicochemical nature of the API.
Table 1.1 Summarises Comparative Features.
|
Solid Form |
Stability |
Impact on Solubility |
Limitations |
Representative Examples |
|
Polymorphs |
Variable (meta- or stable) |
Depends on form |
Possible transitions |
Ritonavir, Carbamazepine |
|
Salts |
High (often hygroscopic) |
Usually higher |
Needs ionizable site |
Amlodipine besylate, Sertraline HCl |
|
Hydrates/ Solvates |
Often unstable upon drying |
Hydrates ↓ solubility; solvates variable |
Solvent loss risk |
Amoxicillin trihydrate, Theophylline monohydrate |
|
Amorphous |
Thermodynamically low |
High apparent solubility |
Recrystallization risk |
Amorphous indomethacin, Atorvastatin |
|
Co-Crystals |
Generally high |
Tunable solubility |
Needs suitable co-former |
Carbamazepine–saccharin, Caffeine–oxalic acid |
1.3 Pharmaceutical Co-Crystals: Definition and Scientific Basis
The U.S. Food and Drug Administration (FDA) define a pharmaceutical cocrystal as “a crystalline material composed of two or more different molecules, typically an Active Pharmaceutical Ingredient (API) and a coformer, in the same crystal lattice, present in a definite stoichiometric ratio, without proton transfer between the components” [18]. Unlike salts, which involve proton transfer between acidic and basic functional groups, cocrystals are neutral, multicomponent crystals in which constituents are held together by non-ionic, non-covalent interactions (commonly hydrogen bonding, but also halogen bonding, π–π stacking, and van der Waals forces) [19, 20].
The scientific basis of cocrystallization is rooted in supramolecular synthons—predictable, recurring patterns of intermolecular interactions that guide molecular self-assembly in the solid state [21]. By selecting an appropriate coformer (pharmaceutically acceptable and interaction-complementary), one can rationally design lattice architectures that modulate physicochemical properties (e.g., solubility, dissolution, mechanical behavior) without changing the API’s covalent structure or pharmacology [22, 23].
Key advantages of pharmaceutical cocrystals include:
Historical perspective. The cocrystal concept predates modern pharmaceutics; the quinhydrone complex (quinone·hydroquinone, 1:1) reported in the 19th century is a classical early example of a donor–acceptor multicomponent crystal [26]. The modern era of pharmaceutical cocrystals emerged with advances in X-ray crystallography, thermanalysis, and computational design, enabling systematic synthon-guided discovery and translation to drug development [21–23].
Today, FDA and EMA explicitly recognize pharmaceutical cocrystals as a viable, distinct approach for optimizing drug performance; both agencies provide guidance on classification and quality expectations for development and filing [18, 19].
1.4 Non-Covalent Interactions in Co-Crystals (Synthon Approach)
Cocrystal formation, stability, and performance are governed by non-covalent interactions between the API and coformer. Although individually weaker than covalent bonds, these interactions control crystal packing, mechanical properties, and dissolution/bioavailability outcomes [21–23].
Synthon approach. Crystal engineering uses recurrent, predictable interaction motifs (synthons) to plan how molecules assemble in the solid state. Two complementary levels are recognized: supramolecular (structural) synthons (intermolecular motifs built via hydrogen bonding, halogen bonding, π–π/CH–π contacts, etc.) and molecular synthons (intramolecular functional-group fragments predisposing certain contacts) [21, 27]. In pharmaceutical cocrystals, a key distinction is between homosynthons (self-complementary, e.g., –COOH···HOOC dimer) and heterosynthons (complementary, e.g., –COOH···CONH–). Because many drugs have multiple donors/acceptors, synthon thinking introduces hierarchy (which motif forms first) and competition (which interactions dominate under given conditions) [21, 27].
Principal interaction types leveraged in co-crystals:
Appropriate synthon selection and coformer choice allow property-driven design of cocrystals (e.g., raise kinetic solubility without sacrificing stability; improve tabletability without creating hygroscopicity), aligning with developability requirements and regulatory expectations [18–20, 22, 23, 25].
1.4.1 Hydrogen Bonding
Hydrogen bonds are among the most fundamental non-covalent interactions, playing a crucial role in stabilizing molecular aggregates and influencing the structure and properties of chemical and biological systems [30, 31]. Although often described as primarily electrostatic, hydrogen bonds possess a dual character, combining electrostatics, partial covalency, van der Waals dispersion, and polarization effects [32]. This hybrid nature renders them stronger than typical dipole–dipole interactions yet weaker than covalent bonds, positioning them as a unique class of directional intermolecular interactions.
Linus Pauling first defined the hydrogen bond as a link where hydrogen is “simultaneously attracted to two atoms,” thus serving as a bridge between them [33]. The IUPAC definition refines this concept: a hydrogen bond is “an attractive interaction between a hydrogen atom attached to an electronegative donor atom (D–H) and another electronegative atom or group of atoms (acceptor, A),” represented as D–H···A [34].
Nature and Energy Profile
Hydrogen bonds display a broad spectrum of strengths, with bond energies typically ranging from 1 to 40 kcal·mol?¹ [31, 35]. Their magnitude and geometry depend on donor and acceptor type, molecular environment, and degree of covalency.
Modern quantum-mechanical analyses reveal that hydrogen bonding is a composite interaction involving four major contributors [32, 35]:
Hence, hydrogen bonding represents a continuum from weakly dispersive to strongly covalent behavior rather than a single, discrete category [35, 36].
Geometrical Parameters
The geometry of a hydrogen bond is defined by three principal parameters—D, the donor–acceptor distance; d, the hydrogen–acceptor distance; and θ, the D–H···A angle [37].
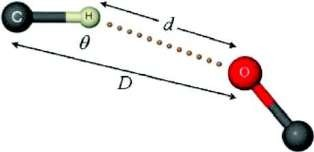
An illustrative schematic of these parameters is shown in Figure 1.2 [39].
Classification of Hydrogen Bonds
Hydrogen bonds are classified as strong, moderate, or weak, based on structural and energetic criteria such as X···A distance, bond angle, and spectroscopic shifts.
This scheme, originally proposed by Jeffrey and Saenger, is summarized in Table 1.2 [36, 38].
Table 1.2 – Classification of Hydrogen Bonds (after Jeffrey, guiding values)
|
Parameter |
Strong |
Moderate |
Weak |
|
Interaction type |
Strongly covalent |
Mostly electrostatic |
Electrostatic/dispersive |
|
H···A bond length [Å] |
1.2–1.5 |
1.5–2.2 |
> 2.2 |
|
X–H bond lengthening [Å] |
0.08–0.25 |
0.02–0.08 |
< 0.02 |
|
X–H vs H···A |
X–H ≈ H···A |
X–H < H···A |
X–H << H···A |
|
X···A distance [Å] |
2.2–2.5 |
2.5–3.2 |
> 3.2 |
|
Directionality |
Highly directional |
Moderate |
Deviation > 20° |
|
Bond angle [°] |
170–180 |
> 130 |
> 90 |
|
Bond energy [kcal·mol?¹] |
15–40 |
4–15 |
< 4 |
|
Relative IR shift ΔνXH [%] |
25% |
10–25% |
< 10% |
|
¹H downfield shift [ppm] |
14–22 |
< 14 |
— |
Significance
The dual electrostatic–covalent nature of hydrogen bonding explains its ubiquity across chemistry, biology, and pharmaceuticals. A modern consensus considers any X–H···A interaction with an energy–distance dependence shallower than r?? to qualify as a hydrogen bond [34].
In crystal engineering, hydrogen bonds dictate molecular packing, solubility, polymorphic stability, and drug–excipient interactions [22, 23, 27, 30]. In pharmaceutical co-crystals, they form the backbone of supramolecular synthons, governing design and predictability.
Examples in co-crystals:
1.4.2 Halogen Bond
Halogen bonding (XB) is a non-covalent, attractive interaction formed between the electrophilic region of a halogen atom (acting as a Lewis acid) and a nucleophilic site (Lewis base) [40]. It is conventionally represented as D···X–Y, where X is the halogen atom participating in the interaction, Y is the atom covalently bound to the halogen, and D denotes the electron donor (Lewis base) [41].
According to the IUPAC definition, a halogen bond is “an attractive interaction between an electrophilic region associated with a halogen atom in a molecular entity and a nucleophilic region in another (or the same) molecular entity” [42]. The interaction is directional, tunable, and highly specific, often compared to hydrogen bonding in strength and geometry.
Types of Halogen Bonds
Halogen···halogen or halogen···nucleophile contacts are typically classified into two types based on geometrical and electrostatic criteria [28]:
σ-Hole Theory
The σ-hole concept, introduced by Politzer and Murray, explains the origin of halogen-bond directionality [44].
Along the extension of the covalent C–X bond, there exists a region of positive electrostatic potential—the σ-hole—that interacts with electron-rich sites such as oxygen, nitrogen, or π-systems [44, 45].
The magnitude of the σ-hole depends on:
Thus, iodine typically forms the strongest halogen bonds, while fluorine seldom participates unless activated by electron-withdrawing groups [46].
Key Characteristics of Halogen Bonds
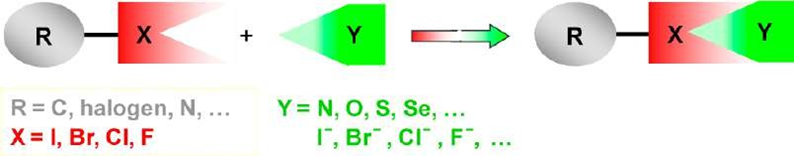
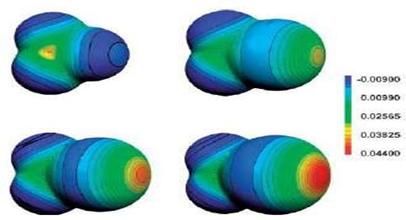
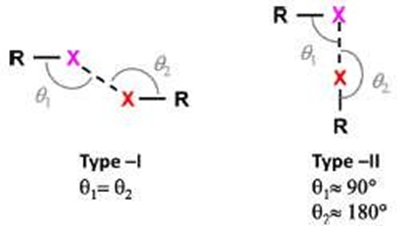
Figure 1.4: (a) Schematic representation of the halogen bond; (b) σ-hole polarization along the C–X axis (increases with decreasing electronegativity); (c) Type I (van der Waals) and Type II (halogen-bonded) X···X or X···Y interactions [44, 46]).
Examples
1.4.3 π–π Stacking Interactions
π-Systems play a pivotal role in supramolecular assemblies due to their capacity to engage in multiple non-covalent interactions dominated by electrostatic and dispersion forces [50]. Among these, π–π stacking interactions are particularly significant in organic crystals, biomolecular structures, and pharmaceutical co-crystals, where they contribute to molecular recognition, packing stabilization, and crystal morphology [51, 52].
Nature and Geometries of π–π Interactions
π–π interactions occur between aromatic systems possessing delocalized π-electron clouds. The interaction geometry and strength are primarily governed by quadrupole-moment complementarity and dispersion stabilization [53]. Two predominant geometries are observed:
The stabilization energy of π–π stacking typically ranges between 2 and 10 kcal mol?¹, depending on aromatic size, substituent effects, and molecular environment [53, 55].
C–H···π (Edge-to-Face) Interactions
Equally important are C–H···π interactions, sometimes called edge-to-face contacts, which originate from a charge-transfer component between a C–H σ-orbital and the π-system [56].
These interactions usually display T-shaped geometry, wherein the positive electrostatic potential of the C–H edge aligns with the negative π-cloud of the aromatic ring.
Two broad categories exist [56, 57]:
C–H···π motifs frequently complement hydrogen-bonding and halogen-bonding networks in the crystal engineering of co-crystals, providing additional stabilization without excessive planarity constraints [58].
Other π-System Interactions
Beyond classical π–π and C–H···π interactions, aromatic systems also engage in:
Together, these diverse π-related interactions provide a toolbox for supramolecular design, influencing mechanical strength, dissolution anisotropy, and electronic communication within functional molecular materials [51, 53].
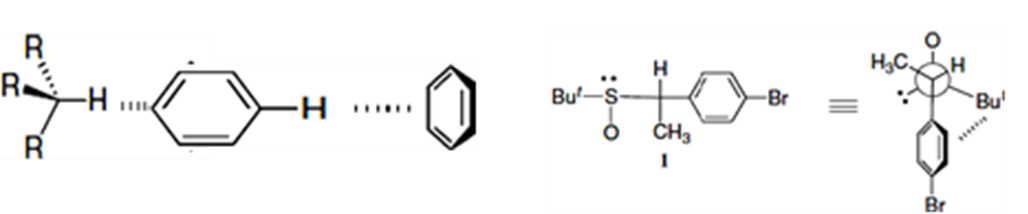
(a) (b)
Figure 1.5: (a) Intermolecular and (b) intramolecular C–H···π interactions [56]).
Examples
1.4.4 Van der Waals Forces
Van der Waals (vdW) forces—named after Johannes Diderik van der Waals, who first introduced the concept of molecular cohesion in 1873—are weak, distance-dependent interactions between atoms, molecules, or surfaces that arise without the formation of covalent or ionic bonds [60].
These forces comprise both attractive and repulsive components and represent the collective outcome of three primary contributions [61]:
Although each component is individually weak (typically < 2 kcal mol?¹), their cumulative effect is essential for cohesion in molecular solids, condensed gases, and organic crystals [62]. In supramolecular systems, vdW forces influence molecular packing, surface adhesion, wettability, and stabilization of crystal lattices [63].
Physical Description
Fluctuating electron densities within atoms or molecules create transient dipoles that synchronize with those of neighboring species, resulting in a dynamic pattern of attractions and repulsions [64].
Quantitatively, the interplay between short-range Pauli repulsion and long-range dispersion attraction defines the overall potential energy curve. This behavior is modeled by the Lennard–Jones (12–6) potential, expressed as:
V(r)=4ε[(σr)12-(σr)6]
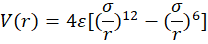
The potential minimum corresponds to the equilibrium separation (≈ 3–4 Å), balancing attractive and repulsive forces [65, 66].
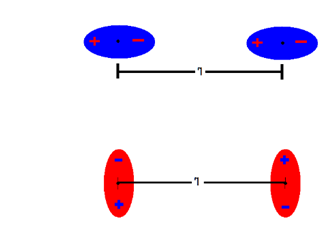
Figure 1.3A illustrates transient electron-density fluctuations responsible for vdW attractions and repulsions,while
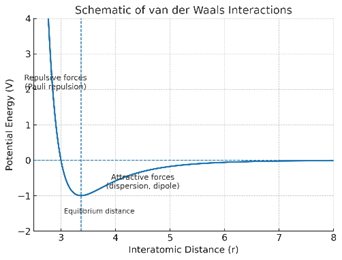
Figure 1.3B depicts the characteristic Lennard–Jones potential curve with the r?? dependence of the attractive term [65, 66]).
Characteristics and Significance
In computational modeling, vdW interactions are incorporated via empirical dispersion corrections (DFT-D) and force-field terms, essential for accurately predicting crystal structures and binding energies [69].
Examples in Co-Crystals
1.5 Methods of Cocrystallisation
The synthesis of pharmaceutical co-crystals can be accomplished through a range of techniques differing in mechanism, operational environment, scalability, and degree of control over crystal quality[7,70].
In general, these techniques fall into three broad categories—solid-state, solution-based, and advanced/emerging methods.
The choice of an appropriate approach depends on the solubility characteristics of the Active Pharmaceutical Ingredient (API) and coformer, thermodynamic stability, desired particle size and morphology, manufacturing feasibility, and regulatory constraints [72].
1.5.1 Solid-State Methods
Solid-state approaches enable co-crystal formation without complete dissolution of the API and coformer, relying instead on mechanical energy, heat, or minimal solvent to facilitate molecular diffusion and intermolecular interaction [6, 74].
These solvent-minimized or solvent-free routes are particularly attractive for green manufacturing and for compounds that are thermolabile or poorly soluble [75].
Neat Grinding (Dry Grinding)
In neat grinding, the API and coformer powders are physically mixed and ground using a mortar and pestle, vibratory mill, or planetary ball mill. The applied mechanical stress induces amorphization, local melting, or defect formation, which promotes hydrogen bonding and lattice rearrangement leading to co-crystal formation [76].
This technique is simple, rapid, and solvent-free, but control over particle size and polymorphism may be limited.
Example: Caffeine–oxalic acid co-crystal synthesized by ball milling without solvent [77].
Liquid-Assisted Grinding (LAG)
LAG involves the same mechanical process as neat grinding but introduces a small quantity (1–10% w/w) of volatile solvent such as ethanol or methanol to enhance molecular mobility and diffusion [78].
Even trace solvent acts as a molecular lubricant, accelerating co-crystal formation and improving reproducibility.
Compared with dry grinding, LAG typically yields higher purity and shorter processing times [79].
Example: Carbamazepine–saccharin co-crystals prepared via ethanol-assisted milling [78, 79].
Polymer-Assisted Grinding
This modification of LAG incorporates polymeric additives such as polyvinylpyrrolidone (PVP) or hydroxypropyl methylcellulose (HPMC), which prevent agglomeration and stabilize metastable or nanosized co-crystal forms [80].
Polymers may also act as nucleation templates, offering additional control over co-crystal morphology and dissolution behavior [81].
Example: Indomethacin–saccharin co-crystals stabilized using PVP during milling [80].
Hot-Melt Extrusion (HME)
HME is a continuous, solvent-free technique where the API and coformer are mixed and heated above their softening or melting points inside a twin-screw extruder, enabling intimate mixing under shear [82].
The molten mixture then cools to crystallize into the desired co-crystal form.
This method offers excellent scalability and process control, aligning with Quality-by-Design (QbD) principles in modern manufacturing [75].
Example: Theophylline–citric acid co-crystals prepared via twin-screw extrusion [82].
Advantages and Limitations
|
Advantages |
Limitations |
|
Solvent-free or minimal solvent use (green method) |
Limited crystal size control |
|
Fast, reproducible, scalable |
Polymorphic control can be challenging |
|
Suitable for heat- and moisture-sensitive drugs |
Mechanochemical heat generation may affect stability |
|
Compatible with continuous processing (HME) |
Process optimization needed for uniformity |
1.5.2 Solution-Based Methods
Solution-based methods rely on the co-dissolution of the API and coformer in a common or mixed solvent system to facilitate molecular self-assembly prior to co-crystal nucleation and growth [23].
Crystallization is then induced by solvent removal, supersaturation, or solubility modulation, enabling the formation of well-ordered crystalline products with controlled morphology [84].
These methods are highly adaptable, provide fine control over crystal size and purity, and are widely used for laboratory screening and industrial scale-up [85].
Solvent Evaporation
In the solvent-evaporation approach, the API and coformer are dissolved in a volatile solvent or solvent mixture in stoichiometric ratio, followed by slow evaporation under ambient or reduced pressure [86].
The gradual increase in supersaturation allows ordered co-crystal nucleation and lattice propagation.
This method is particularly suitable for systems where both components exhibit adequate solubility and thermal stability [87].
Example: Caffeine–glutaric acid co-crystals obtained by slow evaporation from ethanol [86].
Cooling Crystallization
Cooling crystallization involves preparing a saturated hot solution of the API and coformer, which is then cooled gradually to reduce solubility and promote co-crystal nucleation [88].
The temperature-controlled supersaturation minimizes uncontrolled precipitation and allows reproducible particle-size distribution [89].
Example: Paracetamol–oxalic acid co-crystals formed through aqueous cooling crystallization [88].
Anti-Solvent Addition
In this approach, a miscible anti-solvent is added to the solution containing the API and coformer, causing a sudden reduction in solubility that drives rapid co-crystal precipitation [90].
The method is advantageous for APIs with low solubility in non-polar media and enables nanocrystal formation under controlled mixing conditions [91].
However, it requires precise optimization of solvent polarity and addition rate to avoid amorphous or phase-separated products [92].
Example: Naproxen–nicotinamide co-crystals precipitated upon addition of water to an ethanolic solution [90].
Advantages and Considerations
|
Advantages |
Limitations |
|
Produces high-purity, well-defined crystals |
Solvent selection critical for both components |
|
Simple instrumentation; scalable |
Requires solvent-removal steps |
|
Control over morphology and particle size |
Risk of polymorphic variation with solvent type |
|
Suitable for thermolabile APIs (low-temperature operation) |
Potential solvent-residue issues in regulatory compliance |
1.5.3 Advanced and Emerging Methods
Recent progress in crystal engineering and pharmaceutical process design has led to the emergence of energy-efficient and scalable co-crystallisation technologies. These methods offer greater control over particle size, morphology, yield, and phase purity, while aligning with green-chemistry and Quality-by-Design (QbD) principles [6, 94].
Supercritical Fluid Technology
Supercritical fluids, particularly supercritical CO? (scCO?), function as solvents or anti-solvents to generate fine, solvent-free co-crystals [95].
In the supercritical anti-solvent (SAS) process, API and coformer are dissolved in an organic solvent, followed by rapid expansion with scCO?, leading to instantaneous supersaturation and co-crystal precipitation [96].
Example: Ibuprofen–nicotinamide co-crystals synthesized using the SAS technique exhibited uniform micron-sized particles and enhanced dissolution [97].
Ultrasound-Assisted Crystallization
High-frequency ultrasound produces acoustic cavitation, generating localized zones of high pressure and temperature that accelerate nucleation and promote controlled particle-size distribution [98].
Example: Carbamazepine–saccharin co-crystals successfully produced by sono-crystallization [99].
Microwave-Assisted Synthesis
Microwave irradiation provides rapid, volumetric heating, increasing molecular mobility and promoting fast lattice assembly [100].
This method enables co-crystal formation in minutes rather than hours, with minimal solvent and high energy efficiency [101].
Example: Sulfathiazole–salicylic acid co-crystals obtained within minutes under microwave-assisted crystallization [100].
Electrospray Technique
In this technique, a solution containing API and coformer is atomized into charged microdroplets under an electrostatic field, resulting in rapid solvent evaporation and co-crystal particle formation [102].
Example: Indomethacin–saccharin co-crystals generated via electrospray deposition [102].
Laser Irradiation
Focused laser light induces localized heating and melting, followed by rapid recrystallization, offering spatial control over microcrystal formation [103].
Example: Curcumin–oxalic acid microcrystals prepared by laser-induced fusion [103].
Spray Congealing
Here, molten mixtures of API and coformer are atomized into a cooled chamber, resulting in rapid solidification and formation of spherical co-crystalline particles [104].
The method is solvent-free, continuous, and suitable for thermally stable APIs, making it industrially viable for solid dosage manufacturing [94].
Example: Flurbiprofen–saccharin co-crystals produced via spray congealing [104].
Comparative Summary of Co-Crystal Synthesis Methods
Selection of Method for Co-Crystallisation
The choice of co-crystallisation method dictates not only the success of co-crystal formation but also its stability, scale-up potential, and pharmaceutical performance [106].
Since co-crystals form via non-covalent interactions (hydrogen bonding, π–π stacking, van der Waals forces), the chosen technique must ensure intimate molecular contact without compromising chemical integrity [107].
1) Factors Governing Method Selection
2) Common Co-Crystallisation Methods
3) Criteria for Method Selection
|
Criterion |
Preferred Method |
Rationale |
|
API and coformer soluble in same solvent |
Solution-based |
Enables homogeneous nucleation |
|
Thermally unstable API |
Grinding or solvent evaporation |
Avoids heat degradation |
|
Large-scale production |
HME, SCF, spray drying |
Scalable and continuous |
|
Regulatory solvent limits |
Ethanol, isopropanol systems |
Green and pharmaceutically safe |
|
Fine particle size control |
SCF, electrospray |
High precision and tunability |
Selection of Suitable API–Coformer Pairs
The rational design of pharmaceutical co-crystals fundamentally depends on selecting a compatible API–coformer pair capable of favorable non-covalent interactions and pharmaceutical acceptability [106, 107].
1) Molecular and Structural Considerations
APIs containing functional groups such as carboxylic acids, amides, imides, hydroxyls, or heteroaromatic nitrogens readily form directional hydrogen bonds with complementary coformers [108].
Two key supramolecular motifs dominate co-crystal formation: Homosynthons (–COOH···HOOC) and Heterosynthons (–COOH···CONH–, –COOH···pyridine N) [109].
Structural complementarity (shape, size, aromaticity) facilitates packing efficiency and stability through π–π stacking and CH–π interactions [110]. Tools such as the Cambridge Structural Database (CSD) and Hydrogen-Bond Propensity (HBP) models are widely used to predict viable pairs [111].
2) Physicochemical Considerations
The coformer must enhance the solubility, dissolution rate, and stability of the API while maintaining phase integrity under processing and storage.
The ΔpK? rule serves as a useful empirical guideline [106]: ΔpK? < 0 → weak interaction; ΔpK? = 0–3 → ideal for co-crystal; ΔpK? > 4 → salt formation more probable.
Thermal and processing compatibility (melting points, hygroscopicity) must also be evaluated before selecting solvent, grinding, or melt-based methods [108].
3) Regulatory and Biological Considerations
Coformers should have GRAS status and be pharmacologically inert unless therapeutic synergy is desired [112]. Common examples include saccharin, nicotinamide, fumaric acid, tartaric acid, and malic acid [106, 111].
4) Experimental Screening and Validation
Pharmaceutical Examples
|
API–Coformer Pair |
Dominant Synthon |
Observed Improvement |
|
Carbamazepine–Saccharin |
Amide–imide heterosynthon |
↑ Solubility, ↑ dissolution |
|
Caffeine–Oxalic Acid |
Acid–amide heterosynthon |
↑ Stability |
|
Ibuprofen–Nicotinamide |
Acid–pyridine heterosynthon |
↑ Bioavailability |
|
Theophylline–Glutaric Acid |
Acid–heteroaromatic heterosynthon |
↓ Hygroscopicity |
1.6 Applications and Industrial Significance
Pharmaceutical co-crystals have emerged as a versatile solid-state modification strategy offering diverse applications across drug discovery, formulation design, and intellectual property (IP) management [113].
Their industrial significance stems from the ability to tune physicochemical, biopharmaceutical, and mechanical properties of Active Pharmaceutical Ingredients (APIs) without altering the pharmacological structure [109].
1.6.1 Solubility and Bioavailability Enhancement
The most prominent industrial application of co-crystals lies in enhancing the aqueous solubility and dissolution rate of poorly water-soluble APIs—particularly those belonging to Biopharmaceutics Classification System (BCS) Class II and IV [115].
By modifying the crystal lattice through weak intermolecular interactions (e.g., hydrogen bonding, π–π stacking), co-crystals can achieve supersaturated dissolution states, facilitating greater drug absorption [116].
Example: The Febuxostat–arginine co-crystal exhibited a dramatic solubility improvement (571 mg·L?¹) compared with the parent drug (7.5 mg·L?¹), resulting in markedly enhanced dissolution and potential bioavailability [117].
1.6.2 Controlled Drug Release
By rationally selecting coformers with tunable hydrophilicity or hydrophobicity, co-crystals can modulate dissolution kinetics, allowing controlled, delayed, or pulsatile drug release [118].
Example: Theophylline co-crystals with aliphatic dicarboxylic acids demonstrated varying release rates depending on the coformer’s solubility—succinate (fast) vs. adipate (slow) [119].
1.6.3 Multidrug Co-Crystals (MDCs)
Multidrug co-crystals (MDCs) integrate two or more APIs into a single, ordered lattice, maintaining each drug’s chemical integrity while enabling co-delivery and synchronized release [120].
Example: Aspirin–paracetamol co-crystal developed for combined analgesic and antipyretic action demonstrated stable co-forming behavior and uniform dissolution [120].
1.6.4 Mechanical Property Optimization
Poor compressibility and flowability of APIs often hinder direct compression during tablet manufacturing. Co-crystal formation can significantly improve mechanical strength, compactibility, and flow properties by modifying lattice energy and surface morphology [121].
Example: Carbamazepine–saccharin co-crystals showed superior compressibility and flow compared with pure carbamazepine, facilitating robust tableting [121].
1.6.5 Taste Masking
Co-crystallization can effectively mask the bitterness of APIs through complexation with sweet-tasting or neutral coformers, improving patient acceptability without affecting pharmacological activity [122].
Example: Chloramphenicol–saccharin co-crystals improved taste perception while maintaining bioavailability [122].
CONCLUSION
Pharmaceutical co-crystals represent a robust and scientifically grounded solid-state approach for addressing solubility- and stability-related challenges in modern drug development. By harnessing predictable non-covalent intermolecular interactions, co-crystals enable rational modulation of physicochemical and biopharmaceutical properties without chemical modification of the active pharmaceutical ingredient. Advances in crystal engineering principles, screening methodologies, and scalable manufacturing technologies have significantly enhanced the industrial feasibility of co-crystal systems.
With growing regulatory clarity and increasing adoption in pharmaceutical pipelines, co-crystallization is positioned as a valuable alternative to traditional solid-form strategies. Continued integration of co-crystal design within Quality-by-Design and continuous manufacturing frameworks is expected to further expand their role in future pharmaceutical development.
REFERENCES


Anandrao Savkare, Milind Wagh, Pharmaceutical Co-Crystals in Drug Development: Solid-State Design Principles, Intermolecular Interactions, and Manufacturing Strategies, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 4283-4304. https://doi.org/10.5281/zenodo.18110795
 10.5281/zenodo.18110795
10.5281/zenodo.18110795
