Dept. of Pharmacy, Sri Venkateswara College of Pharmacy, Andhra Pradesh.
Cajanus cajan (pigeon pea) is a medicinal plant widely used in traditional medicine due to its rich phytochemical composition and diverse pharmacological activities. The peel, often treated as an agricultural by-product, contains valuable bioactive compounds such as flavonoids, stilbenes, phenolic acids, and terpenoids with potential therapeutic benefits. This study aimed to evaluate the molecular interactions and drug-likeness properties of selected phytoconstituents from Cajanus cajan peel using in silico approaches.Representative compounds including cajaninstilbene acid, orientin, vitexin, genistin, betulinic acid, and phytol were selected based on reported phytochemical data. Their three-dimensional structures were obtained from public databases and optimized prior to analysis. Molecular docking studies were conducted against target proteins associated with inflammation, oxidative stress, and metabolic disorders using validated protocols. Binding affinities, interaction patterns, and key amino acid residues involved in ligand–protein interactions were analyzed to identify potential lead molecules.In addition, pharmacokinetic and toxicity profiles were assessed using ADMET prediction tools, along with drug-likeness evaluation based on Lipinski’s rule of five. The results indicated that several compounds exhibited strong binding affinities and stable interactions within protein active sites. Notably, cajaninstilbene acid and betulinic acid showed promising docking scores and favorable ADMET properties.In conclusion, Cajanus cajan peel-derived phytoconstituents demonstrate significant potential as lead compounds for drug development. However, further in vitro and in vivo studies are necessary to validate these computational findings and confirm their clinical relevance
Pigeon pea (Cajanus cajan), commonly known as toor dal, is a leguminous plant belonging to the Fabaceae family and is widely cultivated in tropical and semi-arid regions [1,2]. It is typically grown as an annual crop and can reach a height of 3–10 feet, characterized by deep root systems, slender leaves, and yellow to reddish flowers [3]. The pods contain 2–9 seeds and serve as an important dietary source in countries such as India, Africa, and Asia. The seeds are nutritionally rich, containing 21–28% protein along with essential vitamins and minerals [4-7]. However, raw seeds contain antinutritional factors such as tannins and trypsin inhibitors, which are significantly reduced upon cooking.
The pods are flat, fibrous, and measure approximately 5–9 cm in length, enclosing hard-coated seeds of varying colors. The peel (pod husk), often discarded as agricultural waste, is fibrous and leathery upon drying, with colors ranging from pale green to purplish brown. Despite being underutilized, the peel represents a potential source of bioactive compounds [8-11].
Phytoconstituents are naturally occurring bioactive compounds synthesized by plants for protection against environmental stress. These compounds exhibit significant pharmacological activities [12,13]. Major classes include phenolics (flavonoids and tannins) known for antioxidant activity, terpenoids with aromatic and antimicrobial properties, alkaloids widely used in therapeutic agents, glycosides with diverse pharmacological actions, and organosulfur compounds with notable biological effects.
In recent years, in silico studies have emerged as powerful tools in drug discovery. These computational approaches utilize models and simulations to analyze biological and chemical systems, predict molecular interactions, and guide experimental studies while reducing time, cost, and animal testing [14]. In silico methods include structure-based approaches such as molecular docking, ligand-based approaches like QSAR, sequence-based analysis, systems biology, and simulation-based techniques such as molecular dynamics.
Molecular docking is a widely used computational technique that predicts the binding orientation and affinity between a ligand and a target protein. It is based on models such as lock-and-key, induced fit, and conformational ensemble [15]. Despite challenges such as protein flexibility and scoring function limitations, docking remains essential in lead identification.
ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction is another critical component of in silico studies, enabling early evaluation of pharmacokinetic and safety profiles [16]. Tools such as SwissADME, pkCSM, ADMETlab, and PASS Online are commonly used for this purpose.
Phytoconstituents of Cajanus Cajan were collected from published research articles and databases such as PubMed, PubChem, and Google Scholar. Reported compounds and their chemical structures were compiled, and structures were downloaded from PubChem in SDF format.
2D structures obtained from PubChem
Converted to 3D using ChemDraw/Chem3D
Energy minimized using MMFF94 force field
Saved in PDB format for docking
Target protein structures were retrieved from the Protein Data Bank (PDB). Selected proteins were prepared for docking by removing unwanted molecules and optimizing structures.
Docking performed using SwissDock
Protein and ligands converted to suitable format
Active site identified and grid set
Binding interactions and energies recorded
Compounds with lowest binding energy considered most active
ADMET properties were predicted using tools like SwissADME, pkCSM, and admetSAR.
Parameters analyzed include:
Absorption: (GI absorption, Lipinski’s rule)
Distribution: (BBB permeability)
Metabolism: (CYP interaction)
Excretion: Toxicity (AMES, hepatotoxicity)
Compounds meeting drug-likeness criteria were identified as potential leads.
3.1 Swiss ADME:
The predicted ADME properties of the phytoconstituents are as provided below:
All the summarized values of Swiss ADME are represented in the table below:
Table-1 Molecular Details of Selected Compounds
|
S.no |
Ligand Name |
GI Absorption |
CYP1A2 Inhibitor |
Lipinski |
Bioavailability Score |
|
1. |
o-Methylisourea hydrogen sulfate
|
Low |
No |
Yes;0 violation |
0.55 |
|
2. |
1,N-dimethyl - N- Propargyl-2-phenylethylminee |
High |
No |
Yes;0 violation |
0.55 |
|
3. |
4,6-Dimethyl-3,5-dioxo-2,3,4,5-tetrahydro-1,2,4-triazine |
High |
No |
Yes;0 violation |
0.55 |
|
4. |
N-[3-Methylaminopropyl] Aziridine |
Low |
No |
Yes;0 violation |
0.55 |
|
5. |
1,3-propanediamine |
High |
No |
Yes;0 violation |
0.55 |
3.2 Molecular Docking:
Molecular docking studies reveal the interaction between proteins and ligands based upon the binding affinity the activity of the compounds was identified. In our project work, we used 15 proteins and 5 ligands i.e. phytoconstituents of Cajanus Cajan when we compared all the 3 compounds, compound 2&3 have most binding affinity
3.2.1 Docking of ligand o-Methylisourea hydrogen sulfate:
1.with protein Casein Kinase II:
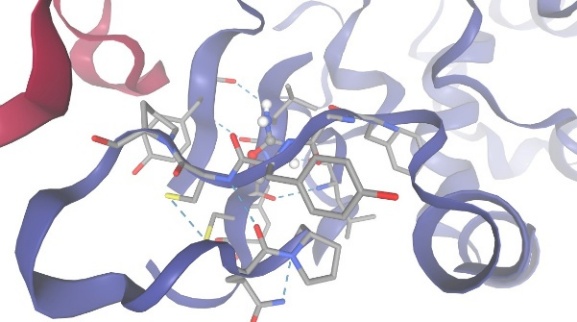
Figure-1 Compound-1 Docking with protein Casein Kinase II
2.With protein Bovine Serum Albumin:
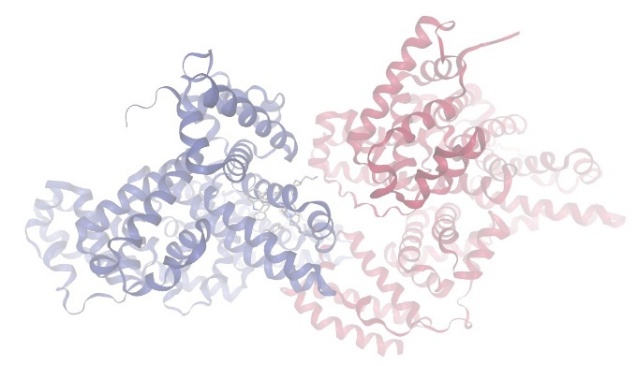
Figure-2 Compound-1 Docking with protein Bovine Serum Albumin
3.With protein Lysozyme:
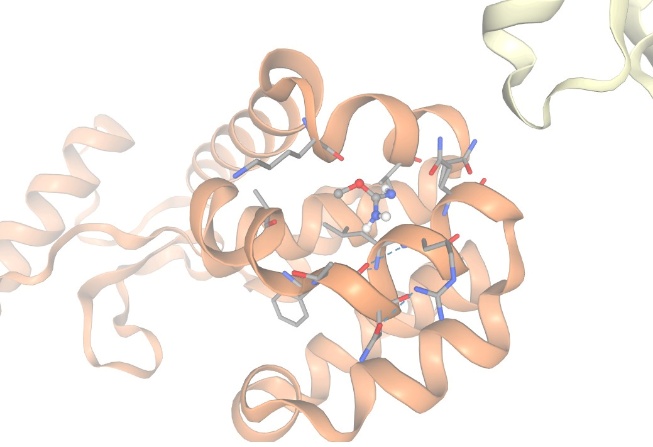
Figure-3 Compound-1 Docking with protein Lysozyme
3.2.2 Docking of 1,N-dimethyl -N- Propargyl-2-phenylethylmine:
1.With protein Human Serum Albumin:
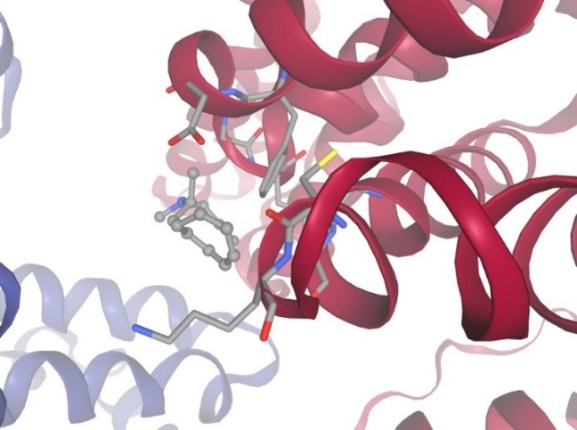
Figure-4 Compound-2 Docking with protein Human Serum Albumin
2.With protein Acetylcholinesterase:
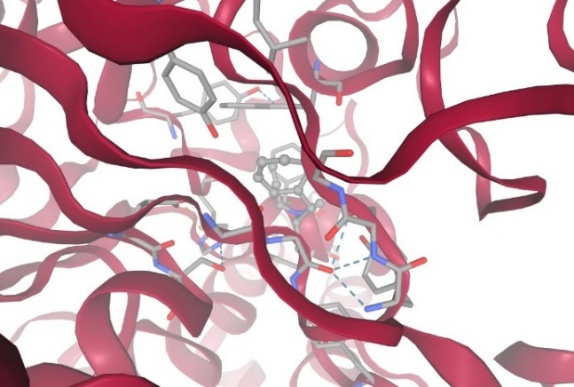
Figure-5 Compound-2 Docking with protein Acetylcholinesterase
3.With protein Bovine Serum Albumin:
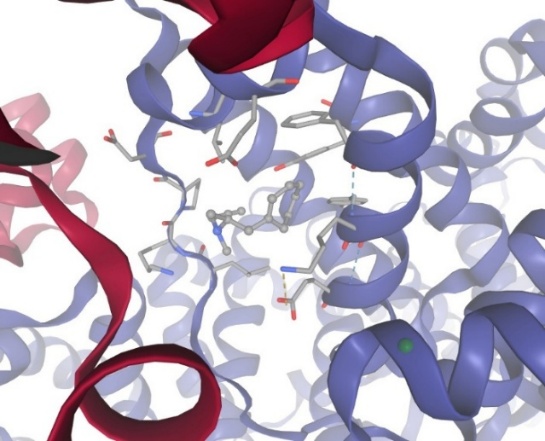
Figure-6 Compound-2 Docking with protein Bovine Serum Albumin
3.2.3 Docking of 4,6-Dimethyl-3,5-Dioxo-2,3,4,5-Tetrahydro-1,2,4-Triazine:
1.With protein Urease:
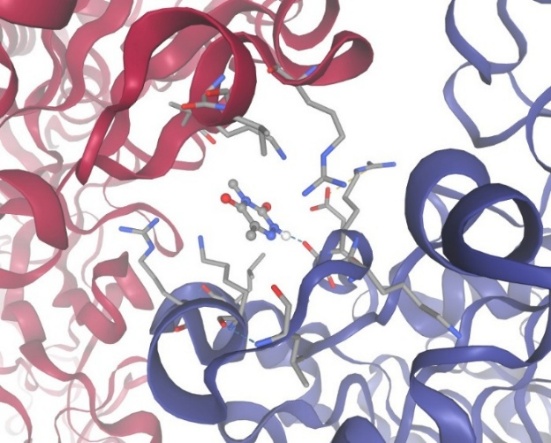
Figure-7 Compound-3 Docking with protein Urease
2.Dihydro Folate Reductase:
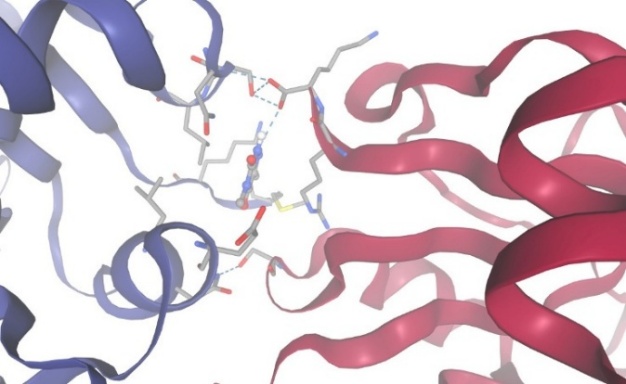
Figure-8 Compound-3 Docking with protein Dihydro Folate Reductase
3.With protein Arginase:
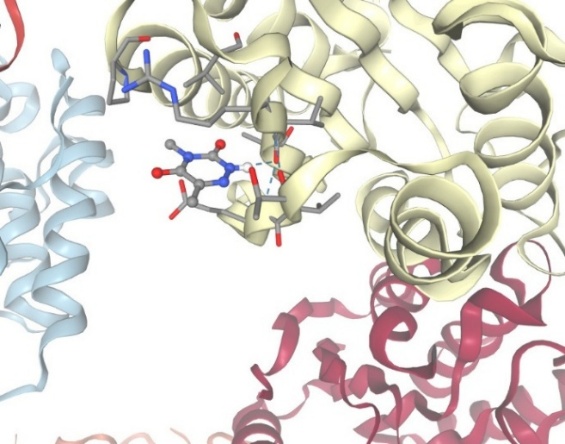
Figure-9 Compound-3 Docking with protein Arginase
3.2.4 Docking of N-[3-Methylaminopropyl] Aziridine:
1.With protein Diamine Oxidase:
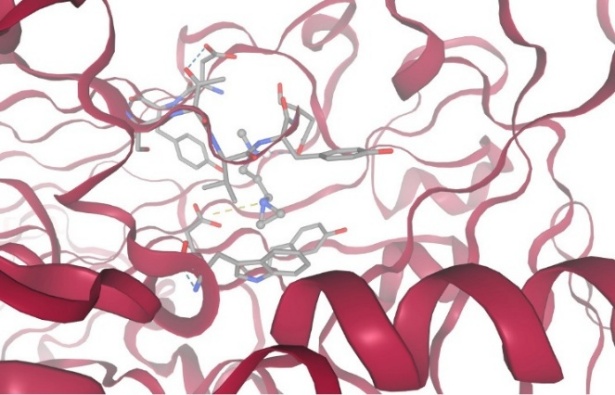
Figure-10 Compound-4 Docking with protein Diamine Oxidase
2.With protein Monoamine Oxidase:
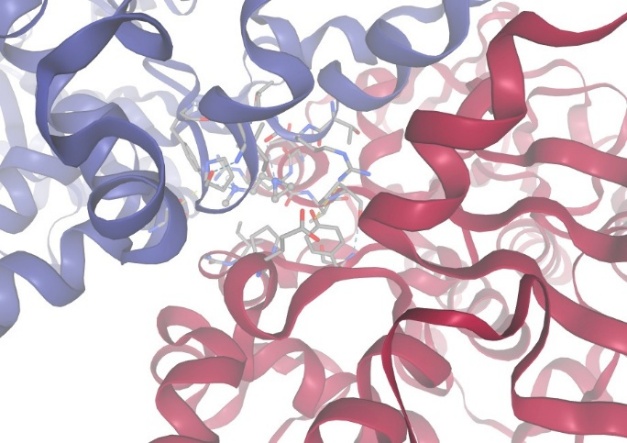
Figure-11 Compound-4 Docking with protein Monoamine Oxidase
3.With protein Dehydrogenase:
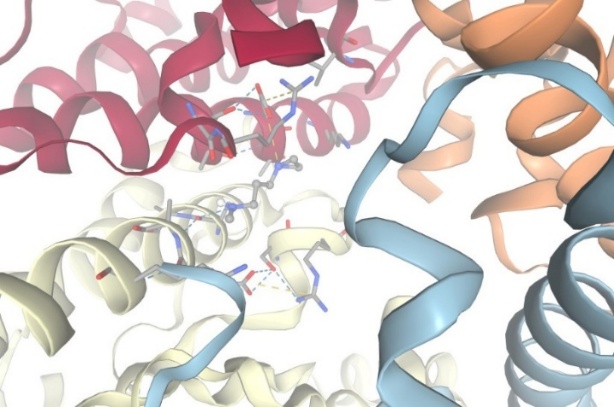
Figure-12 Compound-4 Docking with protein Dehydrogenase
3.2.5 Docking of 1,3-Propanediamine:
1.With protein Diamine Oxidase:
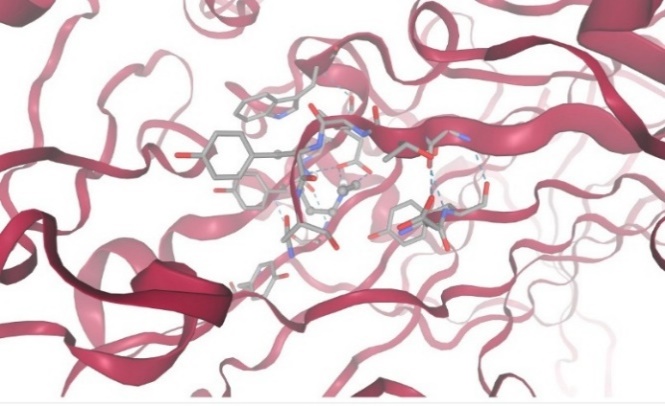
Figure-13 Compound-5 Docking with protein Diamine Oxidase
2.With protein Monoamine Oxidase:
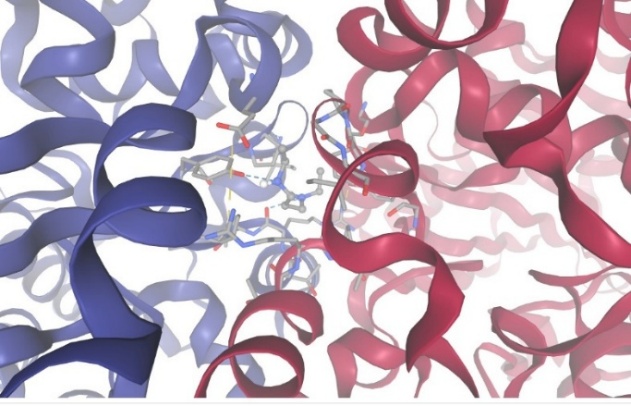
Figure-14 Compound-5 Docking with protein Monoamine Oxidase
3.With protein Polyamide Oxidase:
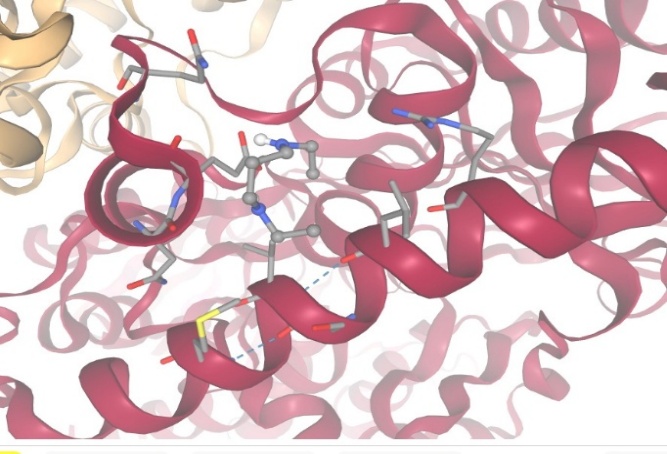
Figure-15 Compound-5 Docking with protein Polyamine Oxidase
3.3 Toxicology Study:
The In Silico toxicology evaluation was carried out to predict the safety profile of the selected phytoconstituents. Compound 2,3,4 are more toxic as they’re showing active toxicity at 4 sites and compound 1 at only one site Computational toxicology prediction tools were used to assess various toxicity parameters such as Hepatotoxicity, mutagenicity, carcinogenicity, cytotoxicity and acute oral toxicity.
Table-2 Toxic activity of the compounds
|
S. No |
Name of the Compound |
Neurotoxicity |
Respiratory Toxicity |
BBB Barrier |
Eco Toxicity |
Clinical Toxicity |
|
1. |
Compound-1 |
Inactive |
Inactive |
Active |
Inactive |
Inactive |
|
2. |
Compound-2 |
Active |
Active |
Active |
Active |
Inactive |
|
3. |
Compound-3 |
Active |
Active |
Active |
Inactive |
Active |
|
4. |
Compound-4 |
Active |
Active |
Active |
Active |
Inactive |
|
5. |
Compound-5 |
Inactive |
Active |
Active |
Inactive |
Inactive |
CONCLUSION
This research focused on evaluating the bioactive phytoconstituents of Cajanus Cajan using computational techniques such as molecular docking, ADMET prediction, and in silico toxicity assessment. These methods provided an efficient platform to understand the interaction of selected compounds with target proteins and to predict their drug-like properties without the need for initial laboratory experimentation.
The docking analysis indicated that certain compounds demonstrated stronger binding interactions with the selected protein targets, suggesting their potential effectiveness as therapeutic agents. In particular, compound-2 and compound-3 showed comparatively better binding affinity, which highlights their suitability as potential lead molecules for further investigation.
Toxicity predictions showed that while some compounds exhibited activity in specific toxicity parameters, others demonstrated relatively safer profiles. This emphasizes the importance of careful selection and modification of compounds to achieve a balance between efficacy and safety.
In summary, the findings of this study suggest that phytoconstituents of Cajanus Cajan have promising potential in drug discovery. Nevertheless, these results are based on computational predictions and must be supported by experimental studies to confirm their biological activity and safety. This work highlights the significance of integrating natural product research with modern computational tools to accelerate the development of novel therapeutic agents.
REFERENCES


Dr. B. Swarupa, Kantipudi Mohana Sudhatri, Karan Kumar, Raushan Kumar, Rupesh Kumar, Buduri Renu, In Silico Study of Selected Phytoconstituents of Cajanus Cajan Peel, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 2558-2566, https://doi.org/10.5281/zenodo.19606519
 10.5281/zenodo.19606519
10.5281/zenodo.19606519
