College of Pharmacy, Imam Abdulrahman Bin Faisal University, P.O. Box 1982, Dammam
Heterocyclic scaffolds form one of the foundations of the anticancer drug development age as these structures are diverse, flexible in their chemistry and their biological activity is exceptional. The introduction of heteroatoms like nitrogen, oxygen, and sulfur into cyclic scaffolds allows the optimal control of physicochemical parameters and molecular interactions, which allows the selective targeting of cancer-associated targets. The review gives an in-depth discussion of the role of heterocyclic compounds in oncology, including molecular design approaches, mechanistic understanding, and clinical translation. The paper talks about the basics of heterocyclic chemistry, such as classification, aromaticity, and the functionality of heteroatoms in biology. The focus is on rational drug designing techniques including molecular hybridization, pharmacophore-based modeling, bioisosteric replacement, and computational techniques that have enhanced the process of developing potent heterocycle-based anticancer agents. Significant types of heterocycles such as systems based on nitrogen, oxygen and sulfur, fused and polycyclic structures and heterocyclic natural products are critically evaluated in terms of their structure and therapeutic importance. Heterocyclic compounds have mechanistic actions against cancer in a wide range of pathways, which are typically complementary, such as cell cycle arrest, apoptosis, kinase inhibition, DNA damage, epigenetic, and angiogenesis/metastasis suppression. The review also presents the clinical relevance of heterocycle-based drugs, an overview of FDA approved drugs, key issues including drug resistance, toxicity, pharmacokinetic issues, and tumor heterogeneity. Lastly, upcoming trends and future directions are discussed and include the incorporation of heterocyclic chemistry with precision oncology, multitarget drug design and highly advanced delivery methods. Overall, this discussion shows the perennial significance of heterocyclic structures in cancer therapeutics and how it can be used to potentially generate more effective and safer anticancer drugs in the future.
Cancer is one of the giants to the overall health of the world with unregulated cell growth, resistance to apoptosis, metabolic restructuring, and invasion and metastasis abilities [1-5]. Although there has been a tremendous improvement in the initial diagnosis, molecular profiling, and treatment, cancer remains a major cause of morbidity and mortality in the world. Complexity and heterogeneity of tumor biology with adaptive resistance mechanisms has restricted the long-term success of most traditional treatment approaches [7-10].
1.1. Global burden of cancer and unmet therapeutic needs
Cancer is one of the major causes of mortality in the world, and as the population grows old, their lifestyles change, people are exposed to environmental conditions and a genetic tendency to this condition, and it is escalating [11]. The World Health Organization estimates that cancer-related death is claimed to be causing deaths in millions of people each year with low and middle income countries paying a larger share of the toll. Lung, breast, colorectal, liver and prostate cancers, among others, are the most common cancers that cause death all over the world despite the progress made in their screening and treatment [12]. Whilst surgery, radiotherapy, and chemotherapy are the cornerstone of cancer management, these treatments have been linked to limited selectivity and large amounts of systemic toxicity. The therapeutic environment has been changed with the introduction of specific therapies and immunotherapies, which have made it possible to modulate oncogenic signaling and immune checkpoints more specifically [13]. Nonetheless, intrinsic or acquired drug resistance, tumor heterogeneity, dose-limiting toxicities, and inappropriate pharmacokinetic activity often impair clinical results [14-16]. The existing treatments are not usually effective to eliminate the disseminated tumor cells or stop the disease recurrence [17]. [18].
1.2. Importance of heterocyclic chemistry in modern drug discovery
The heterocyclic chemistry is a foundation of modern medicinal chemistry, and heterocycles are the structural backbones of an enormous fraction of approved pharmaceuticals. A hetero-cycle is a compound in a ring form where one or more of the ring atoms is a heteroatom, often either nitrogen, oxygen or sulfur [19]. [20]. Heterocycles containing nitrogen, especially, are commonly used because they can undergo important binding with enzymes, receptors and nucleic acids. Heterocyclic compounds that contain oxygen and sulfur further extend chemical space by bringing in special redox, polarity and conformational properties. The pre-eminence of heterocycles in oncology is demonstrated by the fact that diverse anticancer agents that are used clinically are composed of heterocycles [21][23].
1.3. Evolution of heterocyclic scaffolds in anticancer drug development
Heterocyclic compounds have a long history of development in cancer treatment over the last few decades, which has been accompanied by the development of cancer biology, synthetic chemistry, and molecular pharmacology. The early antitumor agents such as classical antimetabolites and DNA-interacting-agents frequently used simple heterocyclic cores to disrupt nucleic acid synthesis or action [24]. An example of the first heterocycle-based drugs to show clinical effectiveness, by inhibiting DNA replication in dividing cancer cells, were pyrimidine and purine analogues. Since the discovery of oncogenic signaling pathways, heterocyclic scaffolds have been growing to be specific to individual molecular drivers of cancer. The discovery of the kinase inhibitors signaled a paradigm shift in the development of anticancer drugs, as heterocyclic cores, including quinazolines, indoles, and pyridines, are the basis of many of the targeted therapies. These compounds take advantage of the ATP-binding sites of the kinases and they are highly specific and offer better therapeutic indices than the conventional cytotoxic agents. New developments in computational methods and synthetic techniques have also contributed to the evolution of heterocyclic scaffolds [25][26].
2. Fundamentals of Heterocyclic Chemistry in Oncology
Heterocyclic chemistry is at the heart of modern medicinal chemistry and it is now of special importance in the design of anticancer therapy. Heterocyclic compounds combine a complex structure with a wide range of functions, which makes the interactions with biological macromolecules related to tumor formation, progression, and metastasis precise. Heterocyclic frameworks offer a platform on which rational drug design can be applied to oncology, in which subtle differences in molecular recognition can spell the difference between success and failure of therapy.[27].
2.1. Definition and classification of heterocyclic compounds
Medicinal chemistry Since heterocyclic compounds may be natural or synthetic. Natural products with anticancer activity, such as alkaloids, antibiotics, and many more, contain heterocyclic cores, which have been the basis of synthetic analogues with better efficacy and safety. The great variety of these classifications highlights the flexibility of heterocyclic chemistry and its ability to meet the multifaceted needs of anticancer drug development.
2.2. Aromatic vs non-aromatic heterocycles
An important difference between heterocyclic compounds is the fact that they are either aromatic or non-aromatic, which significantly affects their chemical stability and their biological activity. Aromatic heterocycles are defined as being cyclically conjugated and electron delocalized, which meet conventional criteria of aromaticity. These compounds have improved thermodynamic stability and a planar structure and these structures frequently permit p-p stacking and other noncovalent interactions with biological targets [31]. Anticancer drugs containing aromatic heterocycles are extremely common because they can be predicted to have an electromagnetic nature and their binding properties are favorable. These molecules have delocalized electrons which enable such interactions with aromatic amino acid residues in proteins and with the bases of nucleic acids. These types of interaction have been especially useful in the inhibition of enzymes that contribute to DNA replication, transcription, and signal transduction pathways that contribute to cancer progression [32]. Alternatively, non-aromatic heterocycles are not fully delocalized and do not have aromatic stabilization. These compounds can be completely saturated or semi unsaturated and can have a higher conformational flexibility than aromatic compounds. Although non-aromatic heterocycles can be less stable, their three dimensional structures can be used to their benefit as a means of occupying unique binding pockets and selectivity to a particular biological target. Aromatic and non-aromatic heterocycles are used complementary in oncology [33] [34].
2.3. Role of heteroatoms (N, O, S) in biological activity
The most common heteroatomic in heterocyclic anticancer agents is nitrogen which is central in molecular recognition. Nitrogen atoms may be hydrogen bond donors or acceptors, take part in ionic interactions and control the molecular basicity. These properties play a crucial role in affinities to enzyme active sites, receptor domains and nucleic acids. Heterocycles containing nitrogen have shown to be particularly useful in targeting kinases and other ATP-dependent enzymes, since they have been demonstrated to form a major interaction with their target enzymes in catalytic pockets and can also mimic native substrates. The heterocycles with oxygen groups help in contributing to polarity and hydrogen-bonding capacity which can affect solubility and membrane permeability [36]. The drug-target complexes can be stabilized by oxygen atoms through the hydrogen bonds and dipole-dipole interaction. Oxygen heterocycles are commonly used in the manufacturing of anticancer drugs to improve pharmacokinetics and tissue distribution. Also, the oxygen atoms can take part in redox reactions, and this can be used to selectively cause oxidative stress in cancerous cells. The heterocycles containing sulfur add a new type of electronic and steric properties that are not present in the counterparts containing nitrogen and oxygen. Sulfur atoms are larger and more polarizable in order to participate in the specific noncovalent interactions, such as chalcogen bonding [37] [38]. The heteroatomic integration of heteroatoms into heterocyclic systems allows the precision of the molecular characteristics that are important in anticancer activity. Medicinal chemists can optimize potency, selectivity, metabolic stability and toxicity systematically by changing the type, number, and position of heteroatoms. This flexibility has enabled heterocyclic chemistry to become an invaluable asset in drug discovery in oncology, in helping bridge the gap between molecular design and clinical therapeutic [39].
3. Molecular design strategies for anticancer heterocycles
Optimization and discovery of heterocycle-based anticancer agents have a great dependence on clear-cut molecular design approaches that combine chemical intuition and biological intuition. Due to the complexity of cancer biology and the adaptive response of the tumor cells, the effective drug candidates should be potent, selective and have good pharmacokinetic properties with minimum toxicity. The current medicinal chemistry deals with these issues by rational drug design, pharmacophore modeling, bioisosteric modification, computational tools and early prediction of absorption, distribution, metabolism, excretion, and toxicity (ADMET). These strategies combined create a unified strategy of the effective development of heterocyclic anticancer therapeutics [40].
3.1. Rational drug design and molecular hybridization
Rational drug design is a structured method of bioactive molecule development, which is founded on the knowledge of disease biology, molecular targets, as well as structure-activity relations. The latter is especially useful within the field of oncology, where there are several well-characterized targets (i.e. a kinase, growth factor receptor, epigenetic enzyme, and DNA-associated protein) that can be used by this strategy. Heterocyclic structures are perfectly designed to be rationally designed as it offers structural rigidity and functional versatile characteristics, as well as can be precisely aligned at the target binding sites [41]. Molecular hybridization is an important aspect of rational drug design that entails the purposeful structuring of two or more pharmacologically significant structural motifs into one molecular construct [42]. Hybrid molecules in the context of anticancer heterocycles are developed to combine the mechanisms of action that complement each other, thus improving the efficacy and minimizing the chances of resistance development [43]. In one instance, a heterocyclic core demonstrated to have kinase inhibitory activity can be conjugated with a different moiety that can alter the apoptosis or oxidative stress signaling. These hybrids have the potential to concomitantly disrupt several processes of oncogenicity, providing a strategic edge over single-target agents. The effectiveness of the molecular hybridization process should rely on the consideration of the linker length, the flexibility, and the orientation since these aspects affect the general conformation and the biological behavior of the molecule [44]. The use of heterocyclic scaffolds as a central platform in hybrid structures is usually attributed to their capacity to retain pharmacophoric functionalities and the expansion capacity of their structures. The rational design and hybridization can therefore be used to produce multifunctional anticancer agents with better therapeutic characteristics [45].
3.2. Pharmacophore-based design approaches
Pharmacophore-based design is a ligand-based design approach which determines the key structural requirements needed to achieve a biological effect, regardless of the chemical scaffold. A typical pharmacophore model has spatially constructed features that include hydrogen bond acceptors and hydrogen bond donors, aromatic rings, hydrophobic centers, and charged centers. These features depict the minimum of the requirements to conduct the interaction with a biological target [46]. Pharmacophore modeling has found application in anticancer drug discovery especially when relatively little structural data is available about the target protein, multiple active ligands are known, or a combination of both. Through studying similarities in active heterocyclic compounds, medicinal chemists can create pharmacophore hypotheses that can help them create new molecules [47-49] [50]. The strategy is useful in maximizing potency, selectivity, and physicochemical properties, and surmounting intellectual property or resistance problems. Pharmacophore modeling can allow the efficient exploration of chemical space and lead identification in oncology, where there are multiple molecular targets of interest, and complex signaling networks are involved [51].
3.3. Bioisosteric replacement in heterocyclic optimization
Bioisosteric replacement is another popular optimization technique of medicinal chemistry where the atom or functional group is substituted by another atom or group with similar physicochemical characteristics but exhibiting better biological or pharmacokinetic properties. Bioisosterism is also important in optimization of lead compounds in heterocyclic anticancer agents to increase efficacy, selectivity and safety [52]. The heterocyclic bioisosteres are especially useful as they permit the gentle control of the electronic distribution, polarity and steric profile without affecting the overall structure of the molecule. As an example, introducing oxygen or sulfur in place of a nitrogen atom into a heterocycle can cause major changes to hydrogen-bonding characteristics and metabolic stability. These alterations can minimize off-target effects or enhance enzyme resistance [53] [54-56].
3.4. Role of computational tools and molecular docking
Computational aids have already been added to the molecular design of anticancer heterocycles, providing information that supplements experimental methods. Computational chemistry, molecular modeling and bioinformatics have allowed the rapid screening of large libraries of compounds, making drug discovery more cost effective [57]. One of the commonest computational techniques in anticancer drug design is molecular docking. It is the forecast of the orientation of a heterocyclic ligand that is desired to occupy in the binding site of a target protein and the strength of the interaction [58]. The docking studies assist in recognizing the important binding interactions, including hydrogen bonds, hydrophobic contacts, and attractions, that inform the process of structural optimization [59]. In addition to docking, molecular dynamics simulations can be used to understand how drug-target complexes are stable or flexible over time, which can also be used to provide an insight into the binding kinetics and conformational changes. Models of quantitative structure-activity relationship also help in correlating chemical properties with biological activity so that rational prioritization of lead compounds can be made. Computational methods in oncology Computational methods have also found particular use in designing heterocyclic kinase and other enzyme active site inhibitors. The identification of multitarget agents support and prediction of resistance-associated mutations are also supported with these tools. Medicinal chemists would be able to make informed decisions by incorporating computational processes in the design process, which would increase their chances of clinical success [60].
3.5. ADMET considerations in heterocycle-based anticancer agents
Although potency and target selectivity are, in fact, the key qualities of anticancer drugs, desirable ADMET properties are equally important to achieve effective clinical translation. A lot of good heterocyclic products still fail in development because of low absorption, metabolism or unacceptable toxicity [61] [62-65].
4. Nitrogen containing heterocycles in cancer therapy
The most notable and the most studied group of scaffold in the field of anticancer drug discovery is that of nitrogen-containing heterocycles. The abundance of the nitrogen atoms in the framework of heterocyclic structures allows their various forms of interaction with biological targets, such as hydrogen bonding, ionic interactions, p-p stacking, and metal ion binding. It is these properties that enable the nitrogen heterocycles to regulate enzymes, receptors, nucleic acids, and signaling proteins that are key to oncogenesis and tumor progression [66]. This means that a significant percentage of clinically approved small-molecule anticancer agents use nitrogen-based heterocyclic cores. Nitrogen heterocycles possess the ability to be tuned in terms of basicity, electronic distribution, and conformational flexibility and are useful in a wide range of biology. These characteristics allow precise manipulation of pharmacodynamic and pharmacokinetic behaviour and such scaffolds are especially good at targeting complex and adaptive disease pathways. This section brings out the key categories of nitrogen-containing heterocycles used in treatment of cancer with special references to their structures, mechanisms of action and their therapeutic importance [67-69].
4.1. Pyridine and dihydropyridine derivatives
The six-member aromatic heterocycle pyridine, with one nitrogen atom, is a commonly used heterocycle in the design of anticancer drugs because of its moderately high basicity and good binding qualities [70]. The nitrogen atom of pyridine is a hydrogen bond acceptor and is involved in electronic stabilization and helps in the association between the molecule and the active sites of the enzyme and receptor domains. Kinase inhibitors, DNA-binding agents and signal transduction pathway modulators often have pyridine derivatives to enhance solubility, stability and activity. Compounds with pyridines have also shown effectiveness in the field of oncology by stopping cell growth, promoting apoptosis, and interrupting angiogenesis [71][74].
4.2. Pyrimidines and purines as antimetabolites
Some of the earliest and most successful nitrogen-containing heterocycles used in the treatment of cancer are pyrimidines and purines. These heterocycles can form the backbone structure of nucleic acids and their close similarity with endogenous bases can serve as antimetabolites interfering with cell division through DNA and RNA synthesis in fast growing cancer cells. Central to anticancer chemotherapy are the pyrimidine derivatives, especially by blocking the enzyme of thymidylate synthase, as well as disrupting nucleotide biosynthesis [75] decrease myelosuppression and increase therapeutic indices. However, pyrimidines and purines continue to be the backbones in anticancer chemotherapy [76].
4.3. Quinoline and quinazoline frameworks
Quinoline and quinazoline heterocycles are fused nitrogen-containing systems which have gained significant popularity in targeted cancer therapy. These bicyclic structures are aromatically stable and structurally rigid structure, allowing them to have high-affinity with protein kinases, and other signaling molecules involved in tumor growth and survival. Quinoline analogs possess varied anticancer activities, such as topoisomerase, microtubule dynamics, and signal transduction pathways inhibition [77] [78].
4.4. Indole and indazole-based anticancer agents
Indole is a nitrogen heterocycle, bicyclic pyrrole that is a benzene ring with a pyrrolidine group attached to it. It is considered to be a recognized privileged scaffold in medicinal chemistry as it is found in many bioactive natural products and synthetic pharmaceuticals. Indole -based compounds have a wide range of activities in cancer therapy, such as cell cycle arrest, induction of apoptosis, and angiogenesis inhibition [79] [81, 82].
4.5. Imidazole, triazole, and tetrazole derivatives
Imidazole, triazole, and tetrazole are five-membered nitrogen heterocycles that are very important in the current anticancer drug design. High nitrogen content defines these heterocycles, giving them tremendous hydrogen-binding ability, adjustable basicity and unusual electronics [83]. The imidazole derivatives have been significant in their capacity to bind metal ions as well as interact with the active site of enzymes. Imidazole-based compounds have been studied in the oncology field, as enzyme inhibitors, hypoxia-targeted therapy, and also as regulators of intracellular pH. They are small and polar and are therefore appropriate to attack intracellular enzymes and signaling proteins [84]. Triazoles are prized because of their chemical stability as well as their bioisosteric versatility. Triazoles act as linkers or pharmacologically active moieties, which increase the metabolic stability and affinity [85]. Tetrazoles, in which the number of nitrogen atoms is four, are commonly used as bioisosteres of carboxylic acids that are less toxic with better metabolic stability and membrane permeability. [86].
5. Oxygen-containing heterocycles in oncology
Heterocycles containing oxygen are a significant and varied group of structural motifs in the discovery of anticancer drugs. The existence of oxygen atom in cyclic structures gives them unique electronic and polarities as well as hydrogen-bonding properties that play an important role in molecular recognition and biological functions [87]. Oxygen heterocycles tend to be less basic and more metabolically compatible than nitrogen heterocycles, and may be converted into better safety and tolerability profiles[88].
5.1. Benzofurans and coumarins
Benzofurans are heterocyclic oxygen bicycles that are composed of a fused furan and benzene ring. Benzofurans are axial molecules with a strong interaction with biological macromolecules due to their aromatic nature and planar geometry, hence they are useful scaffolds in the development of anticancer drugs [89]. The derivatives of benzofuran have been reported to exhibit a wide range of anticancer effects such as blockage of tumor cell proliferation, induction of apoptosis, and inhibition of metastasis. Electronic delocalization and hydrogen-bonding is enabled by the oxygen atom in the furan ring, which improves the affinity of binding with enzyme active sites and receptor domains [90]. Benzofuran-based compounds have been reported to regulate important signaling pathways responsible in cancer progressions including those that govern cell survival and angiogenesis. Lipophilicity and target selectivity may be fine-tuned by structural modification of the benzofuran core to enable its further evolution into anticancer leads [91]. Another influential type of oxygen heterocycle is coumarins, which is based on benzopyrone. They are found in nature in large quantities; and are long known to possess a variety of pharmacological properties. In cancer therapy, coumarin analogs have anticancer effects by various mechanisms, such as cell cycle inhibition, regulation of reactive oxygen species and tumor angiogenic interference. The lactone moiety of coumarin structure is extremely important in biological activity as it allows the specific association with enzymes and regulatory proteins. Coumarin is also an excellent template to chemical modification to introduce substituents to increase potency and selectivity. Coumarins may be good candidates of further development as cancer therapy agents due to their relatively favorable safety profiles and natural origin [92].
5.2. Flavones, isoflavones, and chromones
Flavones, isoflavones, and chromones are a closely related group of oxygen containing heterocycles based upon a standard benzopyran framework. These are compounds that are widely present in plants and which have gained significant interest over the years owing to their strong anticancer properties and pleiotropic biological activities [93]. Having a 2-phenyl-4H-chromen-4-one structure, flavones are anticancer agents that are effective via the control of various cellular pathways. They have been known to cause apoptosis, prevent cell proliferation and prevent tumor invasion and metastasis. The presence of different hydroxyl groups allows flavones to react with proteins and nucleic acids by means of hydrogen bond, as well as, antioxidant activity that can affect the survival of cancer cells. The isoflavones are structurally related to flavones in location of the phenyl substituent, a difference that has major implications on the biological behavior of the isoflavones. The isoflavones have been greatly researched over their use in hormone-dependent cancers, in which they can act in modulating the estrogen receptor signaling [94]. The fact that they can modulate gene expression, cell cycle, and apoptotic processes makes them relevant to oncology. The fundamental heterocyclic building block of flavones and isoflavones, chromones, is also an independent anticancer active building block. Kinases, topoisomerases and other enzymes that have been reported to be inhibited by chromine derivatives play a role in the proliferation of cancer cells. The chemical stability and the structural simplicity of chromones predetermine its usefulness in the rational modification and hybridization strategy to boost anticancer efficacy. Together, flavones, isoflavones, and chromones demonstrate the therapeutic potential of oxygen-containing heterocycles obtained in the natural sources. Their multitargeting activities and good safety profiles justify their further application as anti-cancer agents [95].
5.3. Lactones and epoxides in anticancer drug design
Lactones are cyclic ester types of compounds that take centre stage in anticancer drugs design because they are found in various natural products that are of clinical significance. The rigidity and reactivity of the lactone ring assist in interacting with enzymes and regulatory proteins of cancer progression [96]. A number of compounds developed that include lactones exert their anticancer activity by disrupting microtubule dynamics, causing DNA damage, or inducing apoptosis. Biological action of lactones is directly connected with the reactivity of ester group, which may be hydrolyzed or covalently reacted with nucleophilic residues of target proteins. As much as this reactivity has the advantage of augmenting potency, it also demands cautious optimization to prepare efficacy and toxicity [97].
5.4. Natural oxygen heterocycles with anticancer potential
The natural products with oxygen heterocycles have been widely used as a prolific source of anticancer agents in history, and they remain guiding the drug discovery today. These compounds are usually complicated and novel mechanisms of action with complex architectures which are hard to synthesize. They have been useful as anticancer drug templates because of their evolutionary optimization assimilated in their structural diversity [99]. Natural oxygen heterocycles are highly anticancer, and most of them regulate oxidative stress, cell division, and programmed cell death pathways.[100].
6. Sulfur-Containing Heterocyclic Compounds
The use of heterocycles with sulphur is an important and unique category of scaffold in anticancer drug discovery. The presence of sulfur atoms in the heterocyclic structures adds distinct electronic, steric, and physicochemical characteristics that distinguish the compounds with a sulfur atom in the structure than compounds containing nitrogen and oxygen atoms. Sulfur is bigger and more polarizable and can therefore have stronger noncovalent interaction and binding flexibility in biological targets [101]. Such properties enable the sulfur heterocycles to interact with the enzymes, receptors and redox sensitive proteins that are frequently dysregulated in the cancer cells. Sulfur-based heterocycles have been shown to have a diverse biological nature of applications in oncology such as tumor cell growth, apoptosis, redox homeostasis, and signal transduction pathways. They have the capacity to engage in sulfur-specific interactions including chalcogen bonding and thiol reactivity, which further increases their therapeutic potential. The key classes of heterocycles that contain sulfur as discussed here in relation to cancer treatment are their structural characteristic and pharmacological implications [102].
6.1. Thiazoles and benzothiazoles
Thiazoles are five-membered aromatic heterocycles that have sulfur and nitrogen elements. This heteroatomic structure is a dual heteroatomic structure giving them an equal measure of electronic richness and structural rigidity, which render thiazoles very desirable scaffolds in anticancer drug-design. The derivatives of thiazoles have been extensively documented to demonstrate cytotoxic, antiproliferative and apoptosis-inducing properties on diverse cancer cell lines [103]. The thiazole ring sulfur atom is associated with increase in polarizability and lipophilicity, which makes it easy to interact with the hydrophobic sites of the protein binding sites. The nitrogen atom, in turn, allows making hydrogen bonds and electrostatic interactions, enhancing the affinity to the target. Thiazole based drugs have been investigated as kinase, tubulin polymerization and DNA synthesis and repair enzyme inhibitors. They are not particularly large (relatively) and can be easily prepared in the synthetic form, enabling them to be optimized in terms of potency and selectivity [104] [105].
6.2. Thiophenes and thienopyrimidines
The thiophenes are five-membered aromatic heterocycles that have a sulfur atom in them and are structurally analogous to furans and pyrroles. The thiophenes sulfur atom to ensure a higher level of aromatic stability and hydrophobicity which can promote membrane permeability and targeting. Tiophene derivatives have been integrated into many anticancer agents because of their good balance between chemical and biological actions [106]. Thiazide compounds have been used in cancer therapy with a variety of effects such as anti-proliferative effects, anti-apoptotic effects, and anti-inflammatory effects, as well as anti-angiogenic effects. The planar aromatic structure of these compounds allows them to access the active site of enzymes and the receptor domains effectively and the ability to replace different positions of the structure of these compounds facilitates experimental structure-activity relationships [107] [108].
6.3. Sulfonamide-linked heterocycles
The heterocycles containing a sulfonamide functional group take a special place among sulfur-containing anti-cancer agents, since they have a strong level of polarity and the ability to form hydrogen bonds. The sulfonamide group can also interact with enzymes and receptors that promote cancer by means of hydrogen bonds as a donor and acceptor. Sulfonamide linkages are commonly used in the design of anticancer drugs in heterocycle format, both to conjugate bioactive heterocyclic cores, and to confer high binding specificity to a particular molecular target [109] [111].
7. Fused and polycyclic heterocyclic systems
In anticancer drug discovery, fused and polycyclic heterocyclic systems represent a superior group of molecular architectures. These are heterocyclic or carbocyclic rings that are connected by a common atom to create rigid, planar or semi-planar rings and structures of greater structural complexity. These architectures offer defined three dimensional structures which allow robust and selective interactions with biological macromolecules. Fused and polycyclic heterocycles have been especially useful in oncology, in which careful molecular recognition is essential, and have been used to target enzymes, nucleic acids, and tumor-growth and tumor-survival-related signaling proteins [112].[113].
7.1. Tricyclic and tetracyclic heterocycles
The tricyclic and tetracyclic heterocycles are defined as heterocyclic molecules which are built around three or four ring systems and which frequently involve several heteroatoms including nitrogen, oxygen or sulfur [114]. The long ring fuses leading to stiff molecular backbones which limit the conformational flexibility, a property that can dramatically increase target specificity. Such rigidity in anticancer drug development enables the best alignment in the active sites of the enzyme or DNA structure, enhancing binding forces and time. Such polycyclic systems often have a strong biological activity because of their extensive p-conjugated surfaces which allow interaction with aromatic amino acid residues and nucleic acid bases via p-p stacking interactions [115] 116].
7.2. Heterocycle-based kinase inhibitors
Protein kinases are of crucial importance in cancer pathogenesis as they control the proliferation, differentiation, survival, and angiogenesis of cells. Kinase signaling pathway dysregulation is also a characteristic of most malignancies and therefore kinases represent the ideal target of anticancer therapeutics [117]. Kinase inhibitors have been especially notable examples of the success of fused heterocyclic scaffolds because of their capacity to mimic adenosine triphosphate (ATP) and bind kinase active sites specifically. Kinase inhibitors based on heterocycles are usually designed to take advantage of fused nitrogen based systems to match the hinge region of kinase and remain in critical hydrogen bond and hydrophobic contact [118] [120].
7.3. DNA intercalating heterocyclic agents
DNA intercalation is an established anticancer action whereby planar aromatic molecules are inserted between the adjacent base pairs of DNA, which affects its structural integrity and prevents the normal functioning of the cell, including the replication and transcription processes. Heterocycles containing fused and polycyclic compounds are exceptionally favorable to this type of action owing to the plane geometry and the long surfaces of aromatics. The heterocyclic DNA intercalators have anticancer effects due to the ability to cause DNA damage, cause cell cycle arrest, and induce apoptosis [121]. The specificity of binding by heteroatom inclusion in polycyclic systems is increased by hydrogen bonding and electrostatic interactions with the DNA backbone. These reactions stabilize the intercalated complex and extend its effects on nature. Although DNA intercalating agents may be very potent, they are not selective and may also cause toxicity in fast growing normal tissues. As a result, the current drug design approaches are focused on optimizing fused heterocyclic intercalators to enhance tumor selectivity and minimize the adverse effects. It involves the incorporation of substituents that are directed to cancer-specific sequences of DNA or the adoption of prodrug strategies. Regardless of these obstacles, DNA-interacting heterocycles are still a significant constituent of anticancer chemotherapy [122].
7.4. Multitarget-directed heterocyclic frameworks
Single-target therapies are generally not effective due to the complexity and redundancy of cancer signaling networks. This has led to development of multitarget directed ligands, which are attractive approach to oncology. Polycyclic and fused heterocyclic scaffolds are the most suitable in multitarget drug design since it is possible to include several pharmacophoric units in one rigid structure [123]. Multitarget heterocyclic compounds are programmed to activate or inhibit two or more cancer-relevant signaling pathways, including kinase signaling, epigenetics, and apoptosis. The combination of various functional domains of a fused heterocyclic system can help to interact with a variety of biological targets in a coordinated manner, which may minimize the risks of the development of resistance. Designed wise, polycyclic heterocycles offer a stable core onto which strategic functionalization can be done without structural integrity loss. This allows a close regulation of target selectivity and pharmacokinetic activity. Multitarget heterocyclic structures are becoming considered a sensible reaction to tumor heterogeneity and adaptive resistance in anticancer research [124].
8. Heterocyclic natural products and semi-synthetic derivatives
A large fraction of such molecules possess heterocyclic structures, and the main focus of their attachment to cellular targets that cause cancer initiation and progression. The heteroatomic nature of the formed cyclic systems, which are naturally produced, allows focused recognition of the molecule, high-binding specificity and control of a variety of signaling pathways [125].
8.1. Alkaloids as anticancer heterocycles
A significant number of alkaloids are highly cytotoxic and this aspect has led to their extensive application in chemotherapy. Alkaloids may have problems with narrow therapeutic indices and dose-limiting toxicities, although they are effective. These constraints have been the motivation behind major medicinal chemistry efforts directed at structural refinement and controlled delivery. Still, alkaloids are not replaceable heterocyclic structures of oncology and can still be used as models in the design of new anticancer drugs [127].
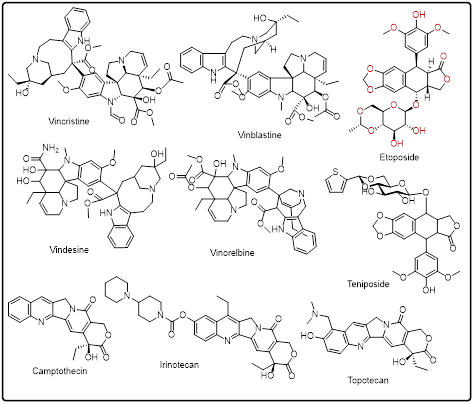
Fig.1: Alkaloid based heterocycles as anticancer agents
8.2. Marine-derived heterocyclic compounds
These characteristics often end up being strong and selective biological activity. Marine heterocyclic compounds have been used in oncology studies, where they have been found to exert anticancer effects in the form of cell cycle arrest, apoptosis, angiogenesis inhibition and signal transduction pathways control [129 [130].
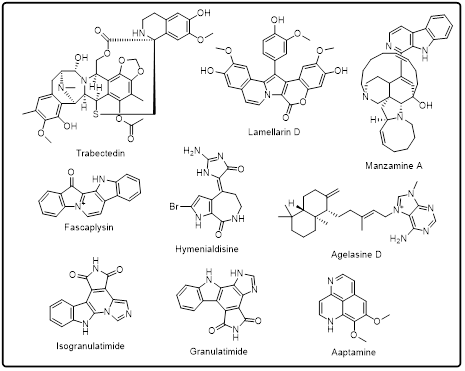
Fig.2: Marine based heterocycles as anticancer agents
8.3. Microbial and plant-based heterocycles
Heterocyclic secondary metabolites that have high anticancer effects are prolifically produced by microorganisms and plants [131] 132], especially those of actinomycetes and fungi. Their effects on cancer are usually associated with the ability to inhibit DNA synthesis, transcriptional interference, and disrupt cellular metabolism. These compounds are heterocyclic, which enhances good interactions with enzymes and nucleic acids, which contribute to their cytotoxic effects. Although numerous heterocycles derived via the plant and microbial route demonstrate good in vitro performance, their application in the clinic is often limited by pharmacokinetic factors. Nevertheless, such natural heterocycles have strong value as lead compounds and as molecular templates to be optimized [133].
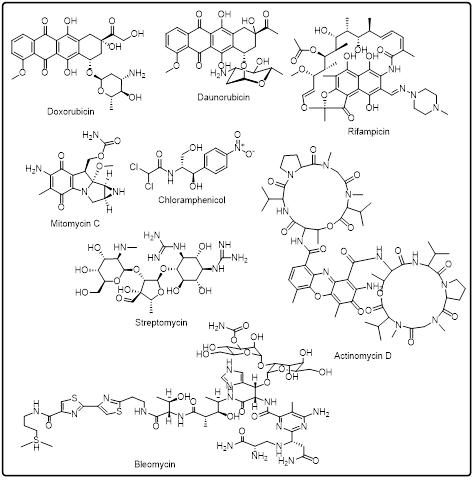
Fig.3: Microbial and plant-based heterocycles as anticancer agents
8.4. Semi-synthetic modification for enhanced efficacy
Semi-synthetic modification is an important approach to enhancing clinical utility of natural products that are heterocyclic. This involves artificial modification of naturally occurring supports through conscious chemical modification to boost potency, selectivity, stability and pharmacokinetic behavior without interfering with the fundamental bioactive framework. Semi-synthetic derivatives have been aggressive in converting promising but inactive natural products into useful anticancer agents in oncology [134].
9. Mechanisms of anticancer action of heterocyclic compounds
Heterocyclic compounds are the structural basis of most modern anticancer agents because of their ability to interact with important molecular targets with high specificity. The integration of the heteroatoms like nitrogen, oxygen and sulfur into cyclic structures provides desirable distribution of electrons, hydrogen-bonding ability and conformational regulation. The attributes allow heterocyclic molecules to regulate various cellular pathways that control tumor formation, growth, survival and spread. In contrast to agents that work using only one mechanism, most agents based on heterocycles have pleiotropic effects, which is especially beneficial in responding to tumor heterogeneity and adaptive resistance. The main anticancer effects related to the heterocyclic compounds will be discussed below [136].
9.1. Cell cycle arrest and apoptosis induction
An unregulated cell division is one of the hall marks of cancer and is a result of cell cycle machinery dysregulation. Heterocyclic compounds often have anticancer effects by inhibiting proteins that regulate cell cycle progression such as cyclins, cyclin-dependent kinases, and checkpoint regulators. Heterocyclic agents disrupt these parts, causing arrest at terminal stages, most often at the G1/S or G2/M phases. This arrest inhibits the replication of DNA or entry into mitosis hence inhibits the growth of tumor cells. A prolonged blockage of cell cycle can result in the stimulation of apoptotic processes. Heterocyclic compounds may induce apoptosis by using intrinsic intrinsic mitochondrial signalling pathways by disrupting mitochondrial membrane potential, facilitating cytochrome c release and caspase cascades. Another way through which they can interact with extrinsic pathways is the approach of regulating death receptors and other similar signaling proteins. Notably, numerous heterocyclic agents, usefully trigger apoptosis exclusively in cancerous cells but not in normal ones, a characteristic due to variations in metabolic status, redox homeostasis as well as apoptotic threshold between malignant and normal tissues. The combination of the two capabilities to prevent proliferation and induce programmed cell death is the basis of therapeutic efficacy of heterocyclic anticancer agents [137].
9.2. Inhibition of kinases and growth factor receptors
Oncogenic signaling is centrally covered by the aberrant activation of protein kinases and growth factor receptors. These enzymes control the pathways involved in cell growth, survival, differentiation, angiogenesis and metastasis. The heterocyclic scaffolds are specifically favorable to the kinase inhibition since these can be able to resemble the endogenous nucleotides and fit perfectly within the ATP-binding pockets or allosteric locations. [138].
9.3. DNA damage and topoisomerase inhibition
Although DNA-targeting heterocyclic compounds can be extremely potent, their clinical application needs a meticulous equilibrium in order to reduce the toxicity to non-cancerous growing tissues. The progress in molecular design and target delivery methods keeps on improving these agents to be more tumor selective and less side effects [139].
9.4. Epigenetic modulation and enzyme inhibition
Epigenetic dysregulation is a serious factor in the development and progression of cancer since it alters gene expression without modifying the DNA sequence. Heterocyclic molecules have been found to be useful as epigenetic enzyme modulators such as histone deacetylases, methyltransferases, and demethylases. Heterocyclic agents are used to inhibit these enzymes and reestablish normal chromatin structure and patterns of gene expression. The control of epigenetics may result in reactivation of tumor suppressor genes, silencing oncogene genes, and cellular differentiation. Notably, epigenetics are reversible and this process is of much interest in long-term therapeutic treatment. [140].
9.5. Anti-angiogenic and anti-metastatic mechanisms
Angiogenesis and invasion of the cancerous cells in the surrounding tissues and colonization in distant organs is what determines tumor growth and metastasis. Heterocyclic compounds have anti-angiogenic actions of inhibiting the endothelial cell proliferation, migration, and endothelial cell generation of new blood vessels, as well as by disrupting the signaling pathways that mediate these effects. [141].
FDA-approved heterocyclic anticancer agents
Heterocyclic compounds have been a decisive element in the development of anticancer therapy in a clinical setting, serving as the structural backbone of a significant percentage of the approved small-molecule drugs. The presence of heteroatoms like nitrogen, oxygen, and sulfur in the cyclic structures complements the best physicochemical characteristics, which allow the high-affinity binding to the cancer-relevant targets. The U.S. Food and Drug Administration gave regulatory approvals that emphasize the core role of heterocyclic scaffolds in modern-day oncology. Antimetabolites, especially those that are pyrimidine-based and purine-based, are some of the earliest heterocyclic anticancer agents that were approved by the FDA [142].
Table 1: FDA-Approved Heterocycle-Based Anticancer Drugs
|
Drug |
Key Heterocyclic Scaffold |
Primary Molecular Target / Mechanism |
Approved Cancer Indications |
|
5-Fluorouracil |
Pyrimidine |
Thymidylate synthase inhibition; DNA/RNA synthesis blockade |
Colorectal, breast, gastric cancers |
|
Capecitabine |
Pyrimidine (prodrug) |
Converted to 5-fluorouracil in tumors |
Colorectal, breast cancer |
|
Methotrexate |
Pteridine |
Dihydrofolate reductase inhibition |
Leukemia, lymphoma, breast cancer |
|
Mercaptopurine |
Purine |
Purine metabolism inhibition |
Acute lymphoblastic leukemia |
|
Fludarabine |
Purine |
DNA polymerase inhibition |
Chronic lymphocytic leukemia |
|
Imatinib |
Pyridine / benzamide heterocycle |
BCR-ABL tyrosine kinase inhibition |
CML, gastrointestinal stromal tumors |
|
Erlotinib |
Quinazoline |
EGFR tyrosine kinase inhibition |
Non-small cell lung cancer |
|
Gefitinib |
Quinazoline |
EGFR tyrosine kinase inhibition |
Non-small cell lung cancer |
|
Lapatinib |
Quinazoline |
EGFR / HER2 inhibition |
Breast cancer |
|
Sunitinib |
Indole |
Multi-kinase inhibition (VEGFR, PDGFR) |
Renal cell carcinoma, GIST |
|
Sorafenib |
Pyridine |
RAF and VEGFR kinase inhibition |
Liver, kidney cancer |
|
Pazopanib |
Indazole |
VEGFR, PDGFR inhibition |
Renal cell carcinoma |
|
Doxorubicin |
Anthracycline (polycyclic heterocycle) |
DNA intercalation, topoisomerase II inhibition |
Breast cancer, leukemia, lymphoma |
|
Daunorubicin |
Anthracycline |
DNA intercalation, topoisomerase II inhibition |
Leukemia |
|
Irinotecan |
Quinoline (camptothecin derivative) |
Topoisomerase I inhibition |
Colorectal cancer |
|
Topotecan |
Quinoline |
Topoisomerase I inhibition |
Ovarian, lung cancer |
|
Vorinostat |
Heterocyclic hydroxamic acid |
Histone deacetylase inhibition |
Cutaneous T-cell lymphoma |
|
Temozolomide |
Imidazotetrazine |
DNA alkylation |
Glioblastoma, melanoma |
|
Thalidomide |
Glutarimide heterocycle |
Anti-angiogenic, immunomodulatory |
Multiple myeloma |
|
Lenalidomide |
Glutarimide heterocycle |
Immunomodulatory, anti-angiogenic |
Multiple myeloma |
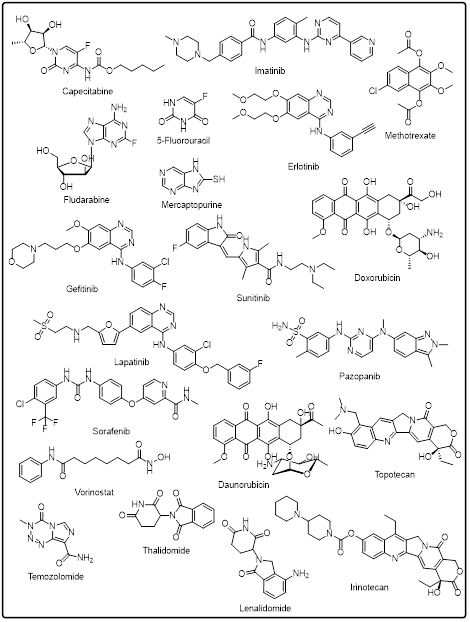
Fig.4: FDA approved heterocycle based anticancer drugs
Challenges and Limitations of Heterocycle based Anticancer Drugs
Although the clinical development and therapeutic use of modern oncology are closely linked to the heterocyclic compounds, they have numerous problems and limitations. Although heterocyclic scaffolds have an outstanding versatility and biological applicability, transition of encouraging preclinical activity to sustained clinical advantage is a multifactorial and complex undertaking. Drug resistance is one of the main issues of the heterocycle-based anticancer treatment. Intrinsic or acquired resistance of cancer cells is often caused by target mutation, redundancy of pathways, or stimulation of reparative pathways. The point mutations at the ATP-binding sites would cause a major decrease in drug affinity in heterocyclic kinase inhibitors, resulting in therapeutic failure. Also, there are other adaptive mechanisms of cellular adaptation like overexpression of efflux transporter and drug metabolism, which further worsen the efficacy of treatment. Another significant limitation is toxicity and off-target effects. Even though, heterocyclic compounds are usually tailored to specifically target a particular target, most of them react with structurally related enzymes or receptors that are also expressed in normal tissues.
CONCLUSION
Heterocyclic structures have proven themselves as essential structural patterns in anticancer drug discovery and development that exist between the concept of molecular design and clinical use. This is due to their inherent chemical flexibility, which is a consequence of the replacement of hydrogen atom centers in cyclic frameworks by heteroatoms, like nitrogen, oxygen and sulfur, which allows the physicochemical and biological properties to be carefully tuned. Heterocyclic compounds serve as the scaffold of many approved anticancer agents and they are still driving the innovation of the traditional chemotherapy paradigm and the targeted therapy paradigm as shown in this review. This attribute can be attributed to the success of heterocycle-based anticancer drugs, reception of them to selectively interact with primary molecular targets that are linked to tumor initiation, progression, and metastasis. These compounds apply therapeutic effects by various mechanisms, such as blockage of cell cycle, induction of apoptosis, inhibition of kinases and growth factor receptors, DNA damage, epigenetic regulation, and inhibition of angiogenesis and metastasis. The ability of heterocyclic scaffolds to support rational design operations like molecular hybridization, pharmacophore modeling as well as bioisosteric modification has further promoted their flexibility towards the intricate and dynamic trajectories of oncogenesis. The heterocycle-based therapeutics with their clinical significance bear significant issues such as drug resistance, off-target toxicity, pharmacokinetic issues, and tumor heterogeneity
REFERENCES


Mohsina Bano Shaik, Saira Zahoor, Heterocyclic Frameworks in Oncology: from Molecular Design to Clinical Application, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 2924-2952. https://doi.org/10.5281/zenodo.18366035
 10.5281/zenodo.18366035
10.5281/zenodo.18366035
