1Bachelor of pharmacy, Rajiv Gandhi Institute of Pharmaceutical Sciences and Research, Trikaripur, Kasaragod, India
2Associate Professor, Department of Pharmaceutical chemistry, Rajiv Gandhi Institute of Pharmaceutical Sciences and Research, Trikaripur, Kasaragod, India.
Curcumin is a bright yellow chemical produced by plants of the Curcuma longa species. It is the principal curcuminoid of turmeric (Curcuma longa), a member of the ginger family, Zingiberaceae. Curcumin incorporates a seven-carbon linker and three major functional groups: an ?, ?- unsaturated ?- diketone moiety and an aromatic O- methoxy - phenolic group. Curcumin has several drawbacks that limit its potential as a therapeutic agent. One of the most common approaches to overcoming these limitations is to design and synthesis some curcumin analogues which contain some heterocyclic moiety. Our aim was to develop novel curcumin derivatives that can provide anticancer activity. In-silico design of novel analogues was carried out using ACD labs chemsketch 12.0. Molinspiration software was used to analyse 'Lipinski rule of 5' and drug likeness properties. Biological activity was predicted by PASS software. ADMET prediction was carried using SWISS ADME software. Preliminary docking study was carried out using PyRx software. The results were compared with standard anticancer drug 5-flurouracil. The results of present project work showed that the novel derivatives of curcumin have comparable anticancer effect with that of standard anticancer drug 5-flurouracil. This will lead to the development of promising lead compounds as anticancer agent and encourage further optimisation to develop potent anticancer drug.
DRUG DISCOVERY PROCESS
Drug discovery is a multifaceted process, which involves identification of a drug chemical therapeutically useful in treating and management of a disease condition. Typically, researchers find out new drugs through new visions into a disease process that permit investigator to design a medicine to stopover or contrary the effects of the disease. The process of drug discovery includes the identification of drug candidates, synthesis, characterization, screening, and assays for therapeutic efficacy. When a molecule avails its satisfactory results in these investigations, it will commence the process of drug development subsequent to clinical trials. Drug discovery and development is an expensive process due to the high budgets of R&D and clinical trials. It takes almost 12-15 years to develop a single new drug molecule from the time it is discovered when it is available in market for treating patients.
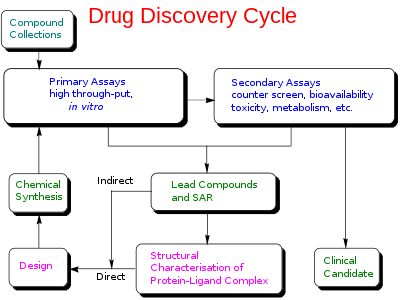
Figure 1: Drug Discovery Cycle
DRUG DESIGN
Design is an integrated developing discipline which portends an era of 'tailored drug'. It involves the study of effects of biologically active compounds on the basis of molecular interactions in terms of molecular structure or its physicochemical properties involved. It studies the processes by which the drug produces their effects, how they react with the protoplasm to elicit a particular pharmacological effect or response how they are modified or detoxified, metabolized or eliminated by the organism. Disposition of drugs in individual region of biosystems is one of the main factors determining the place, mode and intensity of their action. The biological activity may be "positive" as in drug design or "negative" as in toxicology. Thus drug design involves either total innovation of lead or an optimization of already available lead. These concepts are the building stones up on which the edifice of drug design is built up.
STEPS INVOLVED IN DRUG DESIGN
STEP1: identify the structure activity of relationship: The biological effects of a new chemical compound can often be predicted from its molecular structure using data about other similar compounds. This is because similar compounds may have similar physical and biological properties. There is a relationship between molecular structures and their biological activity, and this principle is referred to as Structure Activity Relationship (SAR).
STEP2: identify pharmacophore: A pharmacophore model explains how structurally diverse ligands can bind to a common receptor site. pharmacophore models can be used to identify through de novo design or virtual screening novel ligands that will bind to the same receptor
STEP3: improve target interaction: drug target interaction refers to the binding of a drug to a target location that results in a change in its function. The most common biological targets are nuclear receptors, ion channels, G-protein coupled receptors and enzymes.
STEP4: improve pharmacokinetic properties: by enhancing drug permeation by increasing lipophilicity, or by improving aqueous solubility.
INSILICO DRUG DESIGN
When it comes to rational drug design, the identification and characterization of the biological target of interest is a major step. For the past three decades, drug discovery has been driven by the structure of the target biomolecule. Advances in spectroscopic techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy provided 3D information of many molecules, giving rise to the structure-based drug-design (SBDD) process as a powerful tool for drug discovery in research academia and pharmaceutical industry. In case that the structure of the target protein is unknown, ligand-based computer-aided drug design approaches are used in order to design molecules in silico. Such design is based on information of active or inactive compounds, which already exist and are known that interact with a biomolecular target. The aim of this approach is to retain the important structural and physicochemical properties of the ligand for its interaction with the molecular target while at the same time discard the information which is not related to those interactions.
TYPES OF INSILICO DRUG DESIGN
A. Ligand based drug design: Three main methods are widely used for identifying new molecules with the ligand-based drug-design (LBDD) approach. The first one is called ligand chemical similarity and is based on the selection of new molecules which possess high similarity in chemical structure, binding affinity and physicochemical properties with known active compounds. The second one is pharmacophore mapping, a technique used to identify the functional groups that interact with the targets so as to use this information to design more potent molecules and the last method is quantitative structure- of activity relationship (QSAR), which is used to correlate a number of features from a set chemical compound with their biologic activity for the studied target.
B. STRUCTURE BASED DRUG DESIGN: In the entire drug discovery paradigm, SBDD is the most powerful and efficient process. Computational resources serve as an efficient technology for accelerating the drug discovery process, which includes various screening procedures, combinatorial chemistry, and calculations of such properties as absorption, distribution, metabolism, excretion and toxicity. SBDD is an iterative process and it proceeds through multiple cycles leading an optimized drug candidate to clinical trials.
CHOICE OF LEAD MOLECULE
The lead compound that we have selected for our project work is CURCUMIN.
Curcumin is bright yellow chemical produced by plants of the Curcuma longa species. It is the principal curcuminoid of turmeric (Curcuma longa), a member of the ginger family, Zingiberaceae. It is sold as an herbal supplement, cosmetics ingredients, food flavouring, and food colouring. Curcumin can hold in the management of oxidative and inflammatory conditions, metabolic syndrome, arthritis, anxiety, and hyperlipidaemia. It may also help in the management of exercise-induced inflammation and muscle soreness, thus enhancing recovery and subsequent performance in active people.

Figure 2: Curcumin
Chemically, curcumin is a diarylheptanoid, belonging to the group of curcuminoids, which are phenolic pigments responsible for the yellow colour of turmeric.
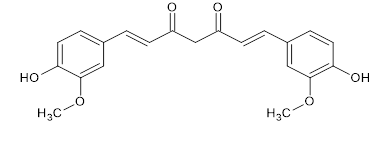
Figure 3: Chemical Structure of Curcumin
Curcumin incorporates a seven-carbon linker and 3 major functional groups: an a, unsaturated b-diketone moiety and a aromatic O-methoxy-phenolic group. The aromatic ring systems, which are phenols, are connected by two ?, ? -unsaturated carbonyl groups. It is a diketone tautomer, existing in enolic form in organic solvents and in keto form in water. The diketones form stable enols and are readily deprotonated to form enolates; the ?, ? -unsaturated carbonyl group is a good Michael acceptor and undergoes nucleophilic addition. Because of its hydrophobic nature, curcumin is poorly soluble in water but is easily soluble organic solvents. Curcumin is used assay complexometric indicator for boron. It reacts with boric acid to form a red -coloured compound, rosocyanine. Cancer is a group of diseases involving abnormal cell growth with the potential to invade or spread to other parts of the body. These contrast with benign tumours, which do not spread. Possible sign and symptoms include a lumb, unexplained weight loss, and a change in bowel movements. While these symptoms may indicate cancer, they can also have other causes. Over 100 types of cancers affect humans. Cancer is the one of the primary causes of death in industrialized countries. In recent years, the early diagnosis and increased in therapeutic options has reduced the death rate. However, the growth of drug-resistant cancers necessitates the search for innovative and more effective drugs. It is worth noting that the cancer cell is characterised by deregulated signalling pathways involving proliferation, apoptosis, and angiogenesis. In this scenario, curcumin represents a promising candidate as an effective anticancer drug to be used alone or in combination with other drugs. It affects different signalling pathways and molecular targets involved in the development of several cancers.
REVIEW OF LITERATURE
EXTRACTION
Extraction is the method of removing active constituents from a solid or liquid by means of liquid solvent. In this method wanted components are dissolved by the use of selective solvents known as menstruum and un dissolved part is marc. After the extraction unwanted matter is removed.
DIFFERENT EXTRACTION TECHNIQUES
MACERATION
In this process turmeric is placed in a stopped container with the whole of the solvent and allowed to stand for a period of at least 3 days (3-7days) with frequent agitation until soluble matter is dissolved. The mixture is then strained (through sieves/nets) the marc pressed. And the combined liquids clarified.
SOXHLET EXTRACTION
In this method the finely ground crude drug(turmeric) is placed in a porous bag or” thimble” made of strong filter paper of the Soxhlet- apparatus. The extracting solvent in a flask is heated, and its vapours condense in condenser. The condensed extractant drips in to the thimble containing the crude drug, and extracts by contact. When the level of liquid in chamber rises to the top of siphon tube, the liquid condenses of chamber siphon in to flask. This process is continuous and is carried out until a drop of solvent from the siphon tube does not leave residue when evaporated.
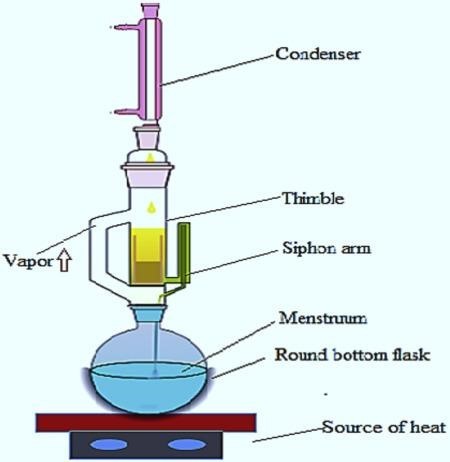
Figure 4: Soxhlet Extraction Apparatus
SOLVENT EXTRACTION
Solvent extraction is a very simple and sensitive technique, useable with a wide range of accelerants. The evidence container is opened and a small quantity (depending on the amount of debris in the container) of a suitable solvent is added. Carbon disulfide is the most popular solvent for this process. The solvent is then poured off and filtered and then evaporated to a small volume leaving behind the accelerant residue. This solution can then be introduced into a gas chromatograph. Disadvantages of solvent extraction are, first, that the solvent will also dissolve unwanted pyrolysis products, matrix materials, and other substances, some of which may interfere with the subsequent analysis and second, that evaporation of the solvent may also cause evaporation of some of the volatile components of the accelerant residues.
ULTASOUND ASSISTED EXTRACTION
In ultrasound-assisted extraction (UAE), mechanical energy, generated by ultrasound waves, is applied to the samples(turmeric). This sonication results in cavitation, the generation of small vacuum bubbles or voids in the liquid, which implode at the solid sample resulting in localized high temperatures (about 4500°C) and pressures (about 50 MPa). These forces produce effects such as sonolysis, destruction of cell membranes, and the extraction of intracellular material.
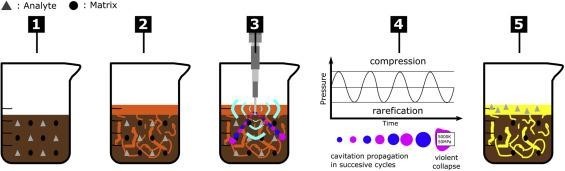
Figure 5: Ultrasound-Assisted Extraction Process
AIM AND PLAN OF WORK
This work aims at the development of novel derivatives containing the versatile lead molecules curcumin. This molecule possesses several therapeutic actions. Aim of the present study design derivatives containing these lead molecules by preforming in-silico studies using different softwares and comparing the anticancer activities of different curcumin derivatives by performing docking studies.
The proposed work was planned as follows:
1. Insilico design
In-silico modelling of molecules using various software such as ACD chemsketch 12.0 is carried out. It is a free software used to draw chemical structures, including organics, organometallics, polymers, and Markush structures, calculate molecular properties (molecular weight, density, molar refractivity, and more) and clean up the structure and view it in 2D and 3D.
2. Docking
Docking is a molecular modelling technique that is used to predict how a protein (enzyme) interacts with small molecules (ligands). The ability of a protein (enzyme) and nucleic acid to interact with small molecules to form a supramolecular complex plays a major role in the dynamics of the protein, which may enhance or inhibit its biological function. The behaviour of small molecules in the binding pockets of target proteins can be described by molecular docking. The method aims to identify correct poses of ligands in the binding pocket of a protein and to predict the affinity between the ligand and the protein. Based on the types of ligands, docking can be classified as:
3.Identification of good anticancer drug among the designed curcumin derivatives.
MODULE 2 QUALITATIVE TEST FOR IDENTIFICATION OF PHYTOCONSTITUENTS
|
Alkaloid |
Dragendroff’s test: Drug solution + dragendroff’s reagent (potassium bismuth iodide)
Mayer’s test: Drug solution + Mayer’s reagent (potassium mercuric iodide)
Wagner’s test: Drug solution + Wagner’s reagent (iodine in potassium iodide solution)
|
Orange red colour
Creamy white precipitates.
Reddish brown precipitate |
|
Glycosides
|
Borntrager test: 10g of drug,5-10 drops HCL. Boil on water bath for 10minutes and filter. Filtrate extract CCL4 or benzene and add equal amount of NH3 solution+ shake.
|
Formation of pink or red colour in ammoniacal layer.
|
|
Haemolysis test: A drop blood on slide was fixed with few drops of saponins solution.
Foam test: To 1g drug add 10-20 ml water shake for few minutes. Formation of froth persist for 60120 seconds.
|
RBC becomes ruptured.
Formation of froth persist for 60120 seconds.
|
|
Liberman’s Burchard test: Alcoholic extract of drug evaporated to dryness and extracted with chloroform add drops of acetic anhydride. Followed by con H2SO4 through sides of test tube. Formation of violet to blue colour ring at junction of two liquid.
Keller Kiliani test: To alcoholic extract of drug add equal volume of water and 5ml strong lead acetate solution shake and filter. Filtrate was extracted with equal volume of chloroform and extract was evaporated to dryness and residue dissolved in 3ml glacial acetic acid. Followed by addition of few drops of ferric chloride solution. Then transferred to test tube containing 2ml con. H2S04 |
Formation of violet to blue colour ring at junction of two liquid.
Reddish brown layer formed which terms to bluish green colour after standing. |
|
TANNINS
|
Gold beater skin test: Gold beater skin is a un tanned fresh skin of an animal and is obtained as membrane from intestine of ox. This membrane treated with HCL, rinsed with distilled water and then placed in tannin solution for 5 minutes. It is followed by washing with distilled water placed in FeSO4 solution.
Phenazone test: To 5 ml aqueous solution of tannin add 5g of Na acid phosphate warm, cool and filter add 2% phenazone to it precipitate.
Gelatin test: To 1% gelatin solution add 10% NaCl. Then 1% solution of tannin add to this solution, tannin cause precipitation of gelatin. |
Tannin imparts brown or dark colour to the skin
All tannins are precipitated as bulky colour precipitate.
Tannin cause precipitation of gelatin. |
|
RESINS
|
|
Red colour obtained by globule indicate the presence of volatile oil.
Red colour indicates presence of volatile oil. |
Table 1 : Qualitative Test for Identification of Phytoconstituents
SEPERATION AND ISOLATION TECHNIQUES
THIN LAYER CHROMATOGRAPHY (TLC)
Thin layer chromatography is a technique used to separate non-volatile mixture. It is performed on a sheet of glass or aluminium foil which is coated with a thin layer of adsorbent. Material usually silica gel, aluminium oxide, cellulose (stationary phase) with a small amount of binder gypsum and water. The prepared layer of adsorbent is known as stationary phase, the resultant plate is dried and activated by heating in an oven for some time at 110°C. The thickness of layer 0.1- 0.25 for analytical purpose and 0.5 -2mm for preparative TLC.
COLUMN CHROMATOGRAPHY
Column chromatography is a separation technique in which components of mixture is separated by using a glass column packed with stationary phase and the liquid mobile phase flowing continuously through the column.
HIGH PERFORMANCE THIN LAYER CHROMATOGRAPHY (HPTLC)
It is a precise automatic form of thin layer chromatography with better and advanced separation efficiency and detection limits and is often excellent alternative to gas chromatography and high- performance liquid chromatography.
HIGH PERFORMANCE LIQUID CHROMATOGRAPHY
High-performance liquid chromatography (HPLC), formerly referred to as high-pressure liquid chromatography, is a technique in analytical chemistry used to separate, identify, and quantify each component in a mixture. It relies on pumps to pass a pressurized liquid solvent containing the sample mixture through a column filled with a solid adsorbent material.
MOLECULAR MODIFICATION OF SELECTED MOLECULES FROM NATURAL SOURCE MAKING USE OF SOFTWARES
STRUCTURE CONSTRUCTION SOFTWARE
Chemsketch
It is an open-source software developed by American chemistry development lab which offers a package for drawing molecules (simple or complex), chemical reactions or molecular models to display in 2D and 3Dstructures.
Utility of chem sketch:
It aids the chemistry teachers at high school/UG or PG level in explaining the students the structure of chemical bonds, the nature of functional groups, and the reaction mechanism.
Utility of chem sketch for teachers
Features of chem sketch:
It has the ability to:
|
Derivatives |
Structure |
|
Smilies Notation |
|
Curcumin |
|
|
COc2cc(CCC(=O) CC(=O) CCc1ccc(O) c(OC) c1) ccc20 |
|
Curcumin pyrimidine |
|
|
COC1=CC(\C=C\C2=CC(\C=C\C3=CC(OC) =C(O) C=C3) =NC(O) =N2) =CC=C10
|
|
Curcumin isoxazole |
|
|
COC1=CC(\C=C\C2=NOC(\C=C\C3=CC(OC)=C(O) C=C3) =C2) =CC=C10 |
|
Curcumin pyrazole |
|
|
COC1=CC(\C=C\C2=NNC(\C=C\C3=CC(OC)=C(O) C=C3) =C2) =CC=C10
|
Table 2 : Structure of Designed Curcumin and curcumin derivatives
MOLECULAR DISCRIPTORS ANALYSIS
|
Compound |
Mol. weight |
Molar volume |
Parachor
|
Surface tension |
Polarizability |
Molar refractivity |
|
Curcumin pyrimidine |
392.411 |
285.5±3.0 |
823.3±4.0 |
69.0±3.0 |
46.07±0.5 |
116.22±0.3 |
|
Curcumin isoxazole |
365.385 |
276.7±0.3 |
768.1±4.0 |
59.3±3.0 |
43.03±0.5 |
108.55±0.3 |
|
Curcumin pyrazole |
364.401 |
272.2±3.0 |
773.7±4.0 |
65.2±3.0 |
43.87±0.5 |
110.68±0.3 |
Table 3: Molecular Descriptors Analysis Data
DRUGS PHYSICOCHEMICAL PROPERTIES
MOLINSPIRATION
It offers broad range of cheminformatics software tools supporting molecule manipulation and processing, including SMILES and SD file conversion, normalization of molecules, generation of tautomer's, molecule fragmentation, calculation of various molecular properties needed in QSAR.
This may be defined as the complex balance of the various molecular properties and structural features which determine whether a particular molecular a similar to that of the known drug.
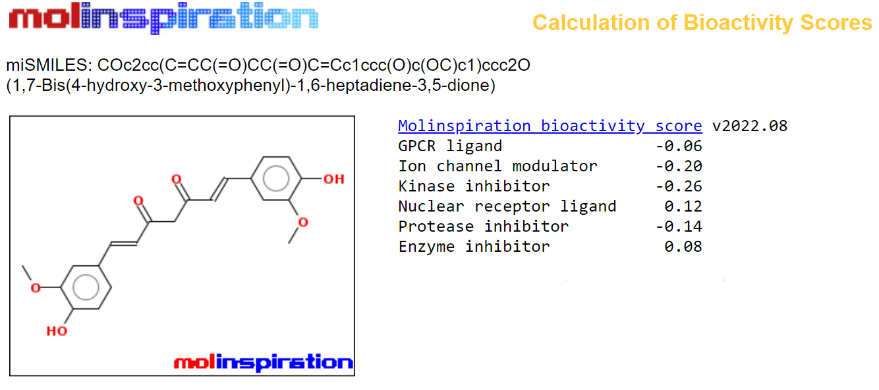
Figure 6: Calculation of Bioactivity Score for Curcumin
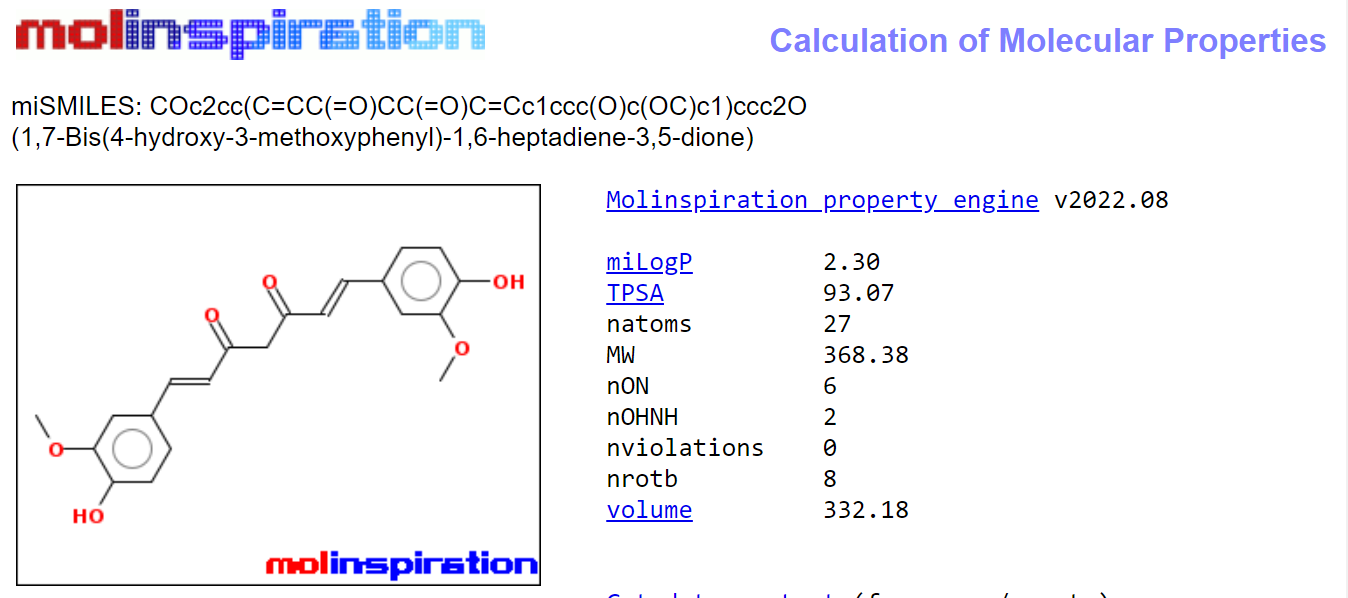
Figure 7: Calculation of Molecular properties of Curcumin
ADMET PREDICTION AND INTERPRETATION
• SWISS ADME
To be effective as a drug, a potent molecule must reach its target in the body in sufficient concentration, and stay there in a bioactive form long enough for the expected biologic events to occur. Drug development involves assessment of absorption, distribution, metabolism and excretion (ADME) increasingly earlier in the discovery process, at a stage when considered compounds are numerous but access to the physical samples is limited. In that context, computer models constitute valid alternatives to experiments and present Swiss ADME web tool that gives free access to a pool of fast yet robust predictive models for physicochemical properties, pharmacokinetics, drug-likeness and medicinal chemistry friendliness, among which in-house proficient methods such as the BOILED-Egg, LOG P and Bioavailability Radar. Easy efficient input and interpretation are ensured thanks to a user-friendly interface through the login-free website http://www.swissadme.ch. Specialists, but also non expert in cheminformatics or computational chemistry can predict rapidly key parameters for a collection of molecules to support their drug discovery endeavours. This web tool used to evaluate pharmacokinetics, drug likeness and medicinal chemistry friendliness of small molecules.
RCSB PDB-RESEARCH COLLABORATORY FOR STRUCTURAL BIOINFORMATICS-PROTEIN DATA BANK
RCSB Protein Data Bank (RCSB PDB) enables breakthroughs in science and education by providing access and tools for exploration, visualization, and analysis of experimentally determined 3D structures from the Protein Data Bank (PDB) archive and computed Structure Models (CSM) from Alpha Fold DB and Model Archive. These data can be explored in context of external annotations providing a structural view of biology. The protein of interest is 7F7W.
Figure 8: Structure of Binding Protein
MODULE 3
PREDICTION OF ACTIVITY SPECTRA FOR SUBSTANCE (PASS)
Pass software is designed as tool for evaluation of general biological potential in the molecule under study. The approach used in PASS is based on the suggestion that activity =f (structure). Thus, by comparing the structure of a new compound with structure of well-known biological active substance it is possible to estimate is a new compound with many thousands of substances from the training set, so provides more objective estimates in a new compound is active or not for any kind of activity as compared with and researcher. PASS training consists of about 60,000 of biologically active compounds. They include about16000 already launched drug and 440000 drug candidates under clinical or advanced preclinical testing. The result of prediction presented as a list of activities with appropriate Pa and Pi sorted in descending order of difference (Pa-Pi) > 0
Pa and Pi are estimates of probability for the compound to be active or inactive respectively foreach type of activity from the biological activity spectrum. Their value varies from 0.000 to 1000
If Pa > 0.7 the compound is very likely to reveal the activity in experiments, but in this case the chance of being the analogue of the known pharmaceutical agents for this compound is also high. If 0.5
CONVENTIONAL SYNTHESIS OF MODIFIED MOLECULES
DERIVATIVES OF CURCUMIN
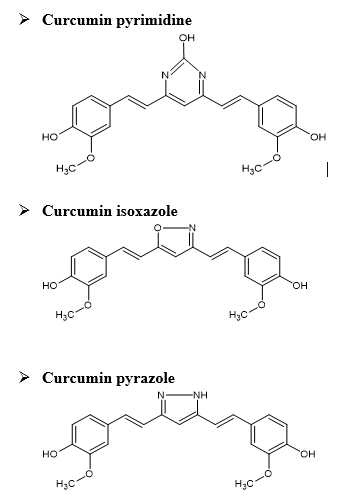
SYNTHESIS OF CURCUMIN ISOXAZOLE
The diketone moiety of curcumin was converted into its isoxazole with hydroxylamine hydrochloride by refluxing in glacial acetic acid for 6-8 hr.
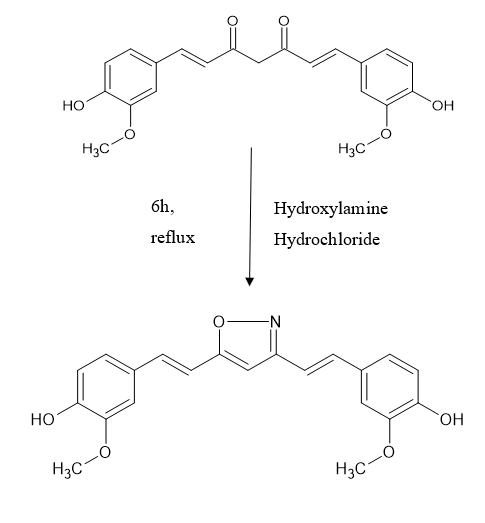
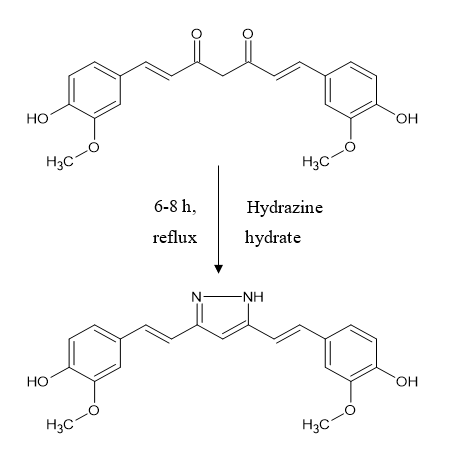
SYNTHESIS OF CURCUMIN PYRAZOLE
Pyrazole derivative of curcumin was synthesized by refluxing curcumin and hydrazine hydrate in glacial acetic acid for 6hr.
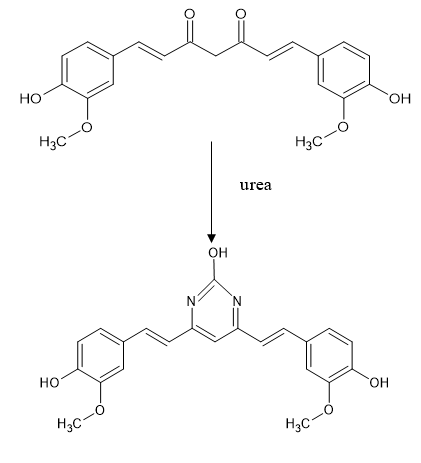
SYNTHESIS OF CURCUMIN PYRIMIDINE
Pyrimidine derivative of curcumin was synthesized by treating with urea.
SCREENING FOR BIOLOGICAL ACTIVITY INSILICO
DOCKING STUDIES
Docking is a method predicts the preferred orientation of one molecule to a second when bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using scoring functions. Molecular docking is one of the most frequently used methods in structure-based drug design, due to its ability to predict the binding-conformation of small molecule ligands to the appropriate target binding site. Characterization of the binding behaviour plays an important role in rational design of drugs as well as to elucidate fundamental biochemical processes.
Two approaches are particularly popular within the molecular docking community;
SHAPE COMPLEMENTARITY
Geometric matching/ shape complementarity methods describe the protein and ligand as a set of features that make them dock able. These features may include molecular surface/complementary surface descriptors. In this case, the receptor’s molecular surface is described in terms of its solvent-accessible surface area and the ligands molecular surface is described in terms of its matching surface description.
SIMULATION
Simulating the docking process is much more complicated. In this approach, the protein and the ligand are separated by some physical distance and the ligands find its position into the protein’s active site after a certain number of ‘moves’ in its conformational space. The moves incorporate rigid body transformations such as translations and rotations, as well as internal changes to the ligands structure including torsion angle rotations.
Figure 9: Docking
DOCKING SOFTWARE
• PyRx
PyRx is a Virtual Screening software for Computational Drug Discovery that can be
used to screen libraries of compounds against potential drug targets. PyRx enables Medicinal Chemists to run Virtual Screening from any platform and helps users in every step of this process - from data preparation to job submission and analysis of the results. While it is true that there is no magic button in the drug discovery process, PyRx includes docking wizard with an easy-to-use user interface which makes it a valuable tool for Computer-Aided Drug Design. PyRx also includes chemical spreadsheet-like functionality and powerful visualization engine that are essential for structure-based drug design.
|
S/no |
Target |
Pd.B. Id |
|
1. |
JAK 2 |
7F7W |
STEPS:
Step 1: Ligand files format conversion
Step 2: Download protein structure
Step 3: Protein structure refinement
Open pymol viewer and load protein. In the command window, give commands as follows:
To extend hydrogen molecules: All action à hydrogen à add file à save molecule ok.
Step 4: Find the grid size
Step 5: Conversion of ligand mol format to pdb format.
Output à pdb format.
Click Convert àclose.
Step 6: Docking with auto dock tools
Close the auto dock.
Step 7: Command line docking:
Search àcmd. Ex
C: /users/m) cd. /…/ (enter click)
C :/> dir
C :/> cd dock
C:/dock) vina-config conf.txt>result.txt
Step 8: Visualizing docked confirmations
MODULE 4
RESULT AND DISCUSSION
RESULT:
The following table shows the physicochemical properties of curcumin derivatives using the software’s;
|
Compound |
Log p |
M W |
nON |
nOHNH |
No.of rotatable bonds |
Violations |
|
5 Fluorouracil |
-0.59 |
130.08 |
4 |
2 |
0 |
0 |
Table 4:Analysis of Lipinski Rule Of Standard Drug
|
compound |
Log p |
M W |
nON |
nOHNH |
nrotb |
violation |
|
CURCUMIN |
2.24 |
372.42 |
6 |
2 |
10 |
0 |
|
CURCUMIN ISOXAZOLE |
3.65 |
371.43 |
6 |
2 |
8 |
0 |
|
CURCUMIN PYRAZOLE |
3.83 |
382.46 |
6 |
3 |
8 |
0 |
|
CURCUMIN PYRIMIDINE |
4.25 |
392.41 |
7 |
3 |
6 |
0 |
Table 5: Analysis of Lipinski Rule of Proposed Derivatives
Molinspiration software was used to analyse Lipinski rule of five and drug likeness properties. From the above tabulated data, it is clear that the derivatives of curcumin obey Lipinski rule of five and it is also clear that there are no violations .so all these derivatives can be used as a promising drug substance.
|
compound |
Log p |
Log s |
GI absorption |
BBB permeation |
Log Kp |
bioavailability |
|
CURCUMIN |
3.03 |
-4.83 |
High |
No |
-6.28 |
0.55 |
|
CURCUMIN ISOXAZOLE |
3.60 |
-5.70 |
High |
No |
-5.54 |
0.55 |
|
CURCUMIN PYRAZOLE |
3.42 |
-5.60 |
High |
No |
-5.64 |
0.55 |
|
CURCUMIN PYRIMIDINE |
3.31 |
-5.93 |
High |
No |
-5.84 |
0.55 |
Table 6: Prediction of Pharmacokinetic Properties by SWISSADME
Using the SWISS ADME software, the pharmacokinetic properties of the proposed derivatives of curcumin was calculated and is mentioned in the above tabular column. From this data it is proved that the curcumin derivatives can be considered as perspective drug candidate.
|
Compound |
Activity |
Pa |
Pi |
|
Curcumin |
Anticancer Anti-inflammatory Antineoplastic |
0,913 0,598 0,874 |
0,003 0,032 0,002 |
|
Curcumin Isoxazole |
Anticancer Anti-inflammatory Antineoplastic |
0,813 0,542 0,679 |
0,007 0,004 0,011 |
|
Curcumin Pyrazole |
Anticancer Anti-inflammatory Antineoplastic |
0,790 0,528 0,761 |
0,009 0,004 0,005 |
|
Curcumin Pyrimidine |
Anticancer Anti-inflammatory Antineoplastic |
0,835 0,339 0,720 |
0,006 0,129 0,002 |
Table 7: Prediction values of various activities using PASS
Using this software various activities of curcumin derivatives such as anticancer, anti-inflammatory and antineoplastic activities are predicted by using this Pa and Pi values.
DOCKING
|
Compound |
Glide Score |
|
Curcumin |
-7.8 |
|
Curcumin Isoxazole |
-8.2 |
|
Curcumin Pyrazole |
-8.1 |
|
Curcumin Pyrimidine |
-8.7 |
Table 8: Docking Scores of Selected Derivatives with Target Protein by Pyrx
From the above tabulated column using the docking software PyRx we have found out that the maximum glide score is shown by curcumin pyrimidine which means that it has better anticancer activity when compared to other derivatives.
SUMMARY AND DISCUSSIONS
The present research work entitled "Golden Spice for the Fight: Unearthing Curcumin's Anti-Cancer Secrets" was concentrated on rational approach to design and develop novel heterocyclic ring substituted curcumin derivatives. The work involved in the preliminary insilico screening of various derivatives to analyse for their molecular descriptors using computational software. The preliminary insilico screening of various derivatives for their drug likeness, ADME profile and analysis of Lipinski rule of five, prediction of biological activities using various software suits like molinspiration, SWISS ADME, PASS, PyRx, etc. Review of literature revealed that curcumin exhibits a versatile biological behaviour and hence it was selected as lead molecule.
The good GLIDE score obtained on docking, highlights the effectiveness of the derivatives for that particular activity. Prediction of ADME properties using SWISS ADME proved that the derivatives can be considered as perspective drug candidates. Derivatives with desired physicochemical properties, obeying Lipinski rule of five and those with no violations were chosen. Evaluation of biological activity was carried to check the anticancer activity of the derivatives and curcumin pyrimidine showed good activity.
The significance of rational drug design approach by means of software’s like PASS, molinspiration, ACD chemsketch, PyRx was useful. The application of insilico studies on designing of drug, molecular docking, drug likeness, prediction of ADME, prediction of biological activity, etc will ensure ecofriendly, economical and time saving methods of drug development with minimum wastage of skill and resources. Further works need to be done in the future for the development of useful chemotherapeutic agents.
REFERENCES





Aswathi K. M.*, Aiswarya lakshmi T., Shahala1, Thanuja ameen K. P., Swathi K. P., Golden Spice for The Fight; Unearthing Curcumin’s Anticancer Secrets, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 1305-1323. https://doi.org/10.5281/zenodo.14375901
 10.5281/zenodo.14375901
10.5281/zenodo.14375901
