1Department of Pharmaceutical Chemistry, Sahyadri college of Pharmacy, Methawde, Sangola
2Associate Professor of Sahyadri college of Pharmacy, Methawde, Sangola
Address for Correspondence: Pradnya Prashant Shinde, Vidyanagar, Akluj road, Malshiras, Dist. Solapur
Effective small molecule development can be achieved through the use of fragment based drug design. Several powerful compounds with a wide range of targets have been created with this method. Finding tiny chemical fragments that bind to biological targets and can then be refined to resemble lead molecules is known as ‘fragment-based drug design’. The methods for identifying fragments, screening them, and turning them into hits or leads—including fragment growth, fragment linking, fragment self-assembly, and targeted libraries—are covered in this article. FBDD will be more crucial to drug development since it is easily conducted using a variety of biophysical and computer-based techniques.
Over the past 15 years, Fragment based drug design has proven to be an effective replacement for high throughput screening in the identification of lead compounds during the drug development process.1 It is a major strategy for identifying high quality lead candidates during past years. 40 compounds identified by FBDD have moved to clinical trials, and two medications already on the market are the result of fragment based library screening efforts.3 This method builds powerful small molecule hits from fragments with minimal molecular mass. For a given protein target, this technique entails creating a diverse fragment library from commercial sources by applying filters (such as natural product-likeness, fraction of SP3 centres, and atom-pair fingerprints-to filter clusters). Next, fragments are affinity screened against the target protein, and fragments are filtered based on affinity numbers. Proteins containing fragments or fragment mixtures are crystallized, and binding sites and modes of fragments are located in three-dimensional structures. These spatial separation and chemical environments of fragments are then used to design linkers that join or expand a fragment. The benefits of biophysical and biochemical methods are used in fragment-based drug design to detect fragments, which are extremely small molecules. The fragment connects to a particular target location. Fragment hits provide an intriguing beginning for the creation of fresh leads. Finding low molecular weight chemical fragments from much smaller compound libraries is the basis of fragment-based drug design. Lead compounds are created by combining or optimizing these pieces. Another name for fragment-based drug design is fragment-based lead discovery. that is employed in the process of discovering new drugs to identify lead compounds.9
What is fragment?
Small organic molecules with a low molecular weight and size are known as fragments. The principle behind FBDD is to find small chemical fragments that bind to biological targets weakly, then expand or combine them to create a lead with a larger affinity. These pieces bind to the target protein in various locations. A lead molecule is created when the fragments mix or link with one another after binding.
The three-rule test determines whether a compound is a fragment.
With the advancement of structural biology and the creation of new screening techniques, FBDD has been easily continued and is now a significant component of target-based drug discovery. To create more active molecules, fragments within protein binding pockets can be expanded, connected, or combined once they have been found.
Identification of fragments
It is challenging to find fragments using the convectional method. Several biophysical screening techniques, like as NMR and X-ray crystallography, are employed in addition to the convectional method. These methods are appropriate since they offer a substantial structural comprehension of the binding event between ligands and proteins. The first real-world application of these concepts was documented by Abbott Laboratories researchers in their "SAR by NMR" description. They made it possible to distinguish between two different molecules that attached themselves to nearby protein surface pockets. A substantial improvement in potency and structural analysis of co-structures found through NMR and X-ray crystallography can be obtained by joining two molecules. It shows that each component of the connected molecule kept its unique binding orientation.2 The following techniques are also used to identify compounds:
Assay for directed or functional binding at high concentration. Generally speaking, fragments have modest binding affinities. They are challenging to find using a standard HTS assay. According to Ellman GR, the way to get around this issue is to filter fragment at a greater concentration.
NMR:
Using NMR screening, one can find tiny molecules that attach to protein targets. a library of fragments that have been filtered to find the ones that attach to the biological targets with N labels. The amide chemical shift that results from compound binding is centred on the nuclear signal quantum correlation spectrum. These data are used with structural details to determine the location of a compound's binding on a protein.10
Crystallography via X-ray
A strong method for obtaining high-resolution protein and complex structures is X-ray crystallography. It is crucial to the structure-based drug discovery process. One reliable technique for identifying and verifying fragment hits is X-ray crystallography. The hardest part of X-ray structural research is getting the targets' and complexes' crystals. Because fragments can be soaked into crystals to determine their binding modes at a high precision, X-ray crystallography is crucial to FBDD. In drug development, X-ray structures are frequently combined with other biophysical techniques. Not every target can be crystallized for X-ray investigations, it has been found. Occasionally, the original fragments soaked into the target crystals may not produce high-resolution structures. Other biophysical techniques must be used in these circumstances to direct fragment growth.
Transforming bits and pieces into hits and leads
Smaller, less complicated molecules with a weaker binder are called fragments. They are more challenging to find using the traditional HTS method. Split often weaker hits acquired through HTS. As a result, they go through a number of procedures to become potential leads. To pursue FBDD, take into account the following actions:
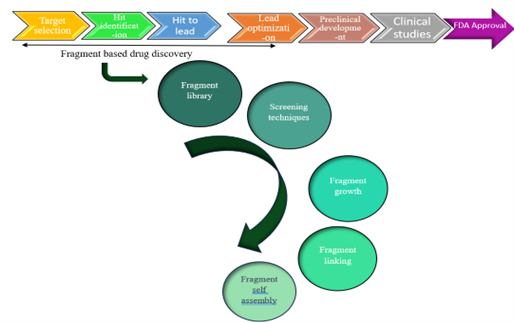
Fig. no 1: Chart shows steps for lead generation
Fragment library:
The quantity and size of compounds in a library are not strictly regulated. The term "fragment" denotes a relatively short molecular weight of the compounds, leading to high ligand efficiency and greater potential for hit growth. Fragments are advised to adhere to the rules-of-three, which state that compounds should have less than three hydrogen donors and acceptors, a molecular weight of less than 300 Da, and a Clog P value of less than three. Typically, researchers use their own specially made FBDD libraries, and a fragment's molecular weight can exceed 300 Da. Because a virtual screening may be completed quickly, the fragment library can be enlarged with an increasing diversity. The quantity of compounds in the fragment library is not a limiting factor because fragments can supply a variety of compounds for optimization. Using FBDD and a library of roughly 800 fragments, a powerful compound was able to be created.
Screening techniques:
Fragments and their targets typically have relatively low binding affinities. It is particularly difficult to investigate fragment and target interactions using biochemical techniques that rely on spectrophotometric and fluorescence assays. Several methods, including differential scanning fluorimetry, x-ray crystallography, and NMR spectroscopy, are employed for the screening process.
Fragment growth:
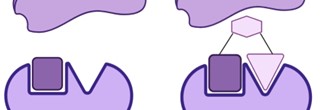
Additional hit optimization is necessary because fragments often bind to targets weakly and don't show strong inhibitory effects on target activity. The first fragment found using the direct binding technique is improved by functionally adding to attach to the active site's surrounding region. The most popular technique for growing fragment into compounds with a higher molecular weight and potency is fragment growing. This picture illustrates how a fragment connects to a receptor in 2(a) and how a lead molecule evolves in 2(b) by growing into a second pocket and expanding away from the beginning fragment and making good contact with the upper surface.
Fragment Linking:
This method is thought to be the most effective for generating strong inhibitors from fragments. It is possible to create a lead compound by joining two or more fragments. The development of chemical binding to protein A replication has been a successful use of this method. Figure 3(a) illustrates how fragment 1 attaches to the receptor at one site, Figure 3(b) illustrates how fragment 2 binds to the receptor at a neighbouring site, and Figure 3(c) illustrates how the connecting group connects the two fragments.4

Fragment self-assembly
The capacity of a fragment to self-assemble when a template is present is known as fragment self-assembly. Another method for transforming fragments into powerful molecules is by fragment self-assembly or merging. This method involves combining the chemical features of two or more fragments to create a powerful molecule. With reactive groups positioned within each other's conformational reach, fragments 1 and 2 bind to the receptor site concurrently in figure 4(a), whereas lead molecule production occurs in the active site in figure 4(b).4
Fig no 4(a) and 4(b)
Targeted libraries:
This strategy optimizes lead's drug-like qualities in addition to binding affinity. The existing lead molecule found using a fragment-based strategy is depicted in Figure 5(a). Additionally, the lead molecule in figure 5(b) has been redesigned to optimize a certain attribute.4
Fig no 5(a) and 5(b)
Developments in FBDD
Universities can undertake fragment-based drug design (FBDD) for tiny biotechnology companies because it is used to evaluate a limited number of compounds. Fragment is chosen to offer perfect beginning points for additional optimization since it interacts with the target protein in an efficient manner. Scientists frequently use x-ray crystallography to study the interaction between the target and fragment as well as to design and synthesize analogs with the necessary desirable features.5 It reduces the level of intricacy. With the recent approval of the covalent KRASG12C inhibitor sotorasib, which highlights the usefulness of FBDD against targets that were long thought to be undruggable, the impact of innovative FBDD techniques is already being seen. FBDD is used in more hydrophilic hits where affinity is more likely to be influenced by hydrogen bonding. Hydrophobic groups make it generally considerably easier to increase affinity. Greater ligand efficiency increases the likelihood that the optimized ligand will have a molecular weight that is quite low (MW < 500>
FBDD's limitations
The variety of protein families that can be targeted by FBDD is one possible drawback. The abundance of high-resolution protein structures is reflected in the numerous instances of kinases and other enzymes as well as PPIs; yet, FBDD has only targeted a small number of membrane proteins. This could soon change, as advances in transmembrane proteins have been made possible by cryo-electron microscopy. Since fragment binding has been demonstrated by the method, FBDD may be used more extensively. As the number of targets and protein families grows, we might be able to determine whether specific binding pocket characteristics are necessary for effective FBDD.13
Prospects for the future of FBDD
Drug leads that come from fragment-based drug design are more likely to be effective. It is anticipated that the fragment strategy would enhance lead compound development quality and productivity in pharmaceutical R&D over the next ten years. The fragmented approach of the future will progress in two directions: first, it will expand and combine fragments into libraries for functional screening, and second, it will break down HTS hits into component fragments for individual optimization. Many of the medications and molecules that FBDD found made it to clinical trials.
CONCLUSION
Drug discovery initiatives should use fragment based drug design. Fragment-based drug design can be applied to a variety of targets, and the fragment screening hit rate can also be used to evaluate the target's drug ability. This information can then be used to direct HTS efforts and provide evidence for a project's approval or rejection. Furthermore, creating strong binders of a protein without enzymatic activity is a great use for FBDD. FBDD has emerged as a credible substitute for large compound library high throughput screens.
REFERENCE:


Pradnya Prashant Shinde, Monika Gopal Shinde, Fragment Based Drug Design: A Review, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 7, 171-176. https://doi.org/10.5281/zenodo.12610829
 10.5281/zenodo.12610829
10.5281/zenodo.12610829
