NRI College of Pharmacy, Pothavarappadu, Agiripalli. 521212.
This study aimed to develop and optimize Mupirocin liposomes for wound healing using a 3^2 full factorial design. The design involved two independent variables, cholesterol and phospholipon 90H, and three dependent variables, percent yield, particle size, and polydispersity index (PDI). The optimized formulation showed a percent yield of 72.87%, particle size of 264.30 nm, and PDI of 0.233. The in vitro release study revealed a diffusion-controlled mechanism, with 95.43% of the drug released within 12 hours. The optimized liposomes were spherical in shape and had a vesicular structure. The study demonstrated the importance of optimizing liposomal formulation for improved wound healing outcomes. The results of this study can be used to develop a topical treatment for wound infections, providing a promising approach for the management of this debilitating condition.
Mupirocin is a potent topical antibiotic used to treat bacterial skin infections, including methicillin-resistant Staphylococcus aureus (MRSA). However, its poor solubility and limited penetration into the skin can hinder its effectiveness. Liposomes, microscopic vesicles composed of lipids, have emerged as a promising drug delivery system to enhance the topical delivery of mupirocin. The present study aims to formulate and evaluate mupirocin-loaded liposomes using Design Expert software, a statistical tool for optimizing formulation parameters. The optimized liposomal formulation will be evaluated for its anti-bacterial property, in vitro drug release, and stability. This study will provide a comprehensive understanding of the potential of liposomes as a topical delivery system for mupirocin, paving the way for the development of more effective treatments for bacterial skin infections. Formulation is a preparation activity in pharmaceutical science that focuses on designing the mix of active components and additives after passing pre formulation research. The traditional formulation step in the development of novel medications employs the strategy of modifying one variable or element at a time. Study the effect of composition and process variables on the dosage form first, then adjust one element while keeping another constant. This traditional method, however, has drawbacks, including being inefficient, unpredictable, time-consuming, and unable to describe the interactions that occur. A new strategy was devised to solve the inadequacies of the traditional formulation process, such as the Experimental Design or experimental design, which is an optimization technique. Optimization is required in the preparation formulation process to establish the optimal formula based on the evaluation data of the prepared product. Under some conditions, optimization can be described as a method for obtaining the best mix of product or process attributes. It can also mean choosing the best element or substance from a variety of possibilities. Various approaches are used to process the data from the predetermined assessment parameters. Software allows for more efficient data processing. Predictive data is combined with data generated after the experiment. Design Expert is one of the most extensively used programs. Expert design is utilized for drug delivery system formulations such as extended-release tablets, targeted drug delivery such as liposomes, ethosomes, and nano particles, in addition to traditional medication formulations such as tablets and capsules. Its applications are not restricted to the pharmaceutical industry; for example, the optimization process for studying the ideal composition of vegetable oil mixtures as raw materials for biodiesel synthesis use Design Expert. Because there are options/features that provide direction and can be selected according to the purpose of the design of experiments (DOE) or experimental design to be carried out, the assisted formulation utilizing the Design Expert is more profitable. As a result, a literature review is required to determine the extent to which Design Experts are used in formulations. The wound is considered one of the complex processes in which the function and integrity of skin and deep underlying tissue are affected, resulting infection and inflammation. Infection of the wound by microbes may frequently occur due to poor hygienic conditions. The penetration and colonization of microorganisms in deeper segments of the tissue may cause tissue damage and, as a result, impede the healing process of the wound. Staphylococcus aureus, Staphylococcus epidermidis, Pseudomonas aeruginosa, Escherichia coli, Streptococcus pyogenes, Klebsiella pneumoniae, and Streptococcus pneumoniae are the most prevalent microorganisms responsible for the infection of wound. A moist environment on the wound bed is favourable to accelerating the cellular growth, migration of epithelial cells, and proliferation of collagen within the noncellular matrix. Therefore, substantial use of antimicrobial agents in wound dressing is required to prevent possible bacterial infection. The most commonly used topical antibiotics are mupirocin, silver sulfadiazine, neomycin, and bacitracin. Mupirocin (MUP, M.W: 500.622 g/mol) exerts antibacterial activity by inhibiting protein synthesis through binding to the isoleucyl-tRNA synthetase of bacteria. MUP is highly active against wound pathogens, e.g., aerobic gram-positive cocci such as Staphylococcus aureus and Staphylococcus epidermidis, as well as some gram-negative cocci, including MRSA. Mupirocin is commercially available as a 2% ointment and treats skin infections, operative wounds, burns, and ulcers.
The solubility of mupirocin is a crucial issue in preparing an aqueous-based formulation. The formation of inclusion complex with water-soluble host molecules is one of the effective ways to improve aqueous solubility. ?-cyclodextrin (M.W: 1135.00 g/mol) is used as a solubility enhancer and is suitable for guest molecules with molecular weight between 200 and 800 g/mol. So far, and to the best of our knowledge, no studies have been done to prepare the mupirocin-?-cyclodextrin (MUP-?-CD) complex in-situ gel. Therefore, the present investigation aimed to enhance the aqueous solubility of MUP by using ?-CD as a complexing agent and to develop MUP-?-CD loaded in-situ gel to achieve a stable drug delivery system. The in-vitro and in-vivo activity of the optimized formulation was compared with a commercial product. The primary goal of wound management is to maintain a clean and moist environment at the wound site to support the physiological healing process, accelerate wound healing and prevent serious infections. Topical therapy is preferred for treating wound infections because the optimal antimicrobial concentration at the wound site for an appropriate duration, facilitates rapid delivery, improves adherence and minimizes the risk of systemic toxicity. However, in many clinical situations conventional topical formulations i.e. creams and ointment, fail to produce the desired therapeutic response because of their inability to adhere at the wound site and lose their rheological characteristics upon contact with the wound fluid. Consequently, non-specific delivery of drugs at infected sites results, necessitating higher doses. Mupirocin (MUP) is a topical antibiotic and drug of choice for treating primary and secondary superficial skin infections caused by SA and multidrug-resistant strains such as MRSA. MUP binds to enzyme the isoleucyl-tRNA synthetase, resulting in RNA and protein synthesis inhibition and ultimately leading to bacterial death. MUP is available on the market in different topical formulations such as creams and ointments. However, their clinical use results in ineffective treatment owing to the poor solubility and dermal penetration of MUP, which necessitate frequent dosing. Nanotechnology offers a potential opportunity to increase the efficacy of current wound therapies by altering the biopharmaceutical and pharmacological characteristics of drugs. Among nanoparticles, stimuli-responsive nanocarriers offer the advantages of targeted and on-demand drug release and can minimize toxicity with improved therapeutic efficacy. Among these stimuli responsive nano-DDS, multifunctional lipid polymeric hybrid nanoparticles (LPHNs) are novel nanocarrier systems composed of phospholipids and polymers. This combination provides advantages of the combined attributes of polymeric nanoparticles and liposomes in terms of increased drug loading capacity, enhanced stability, structural uniformity and rigidity, controlled release, improved biocompatibility, and cellular uptake. For site-specific LPHN-loaded delivery systems the selection of lipids and polymers is important. Chitosan, a natural biodegradable polymer was used owing to its remarkable biological properties such as high stability, non-toxic nature, immunostimulant properties, innate anti-microbial potential and wound healing properties. Chitosan has a positive charge and adsorbs on negatively charged lipid NPs, where it forms a polyelectrolyte complex that release the drug at alkaline pH due to weakening of the complex. Considering the actual need for wound infection, this work was designed to fabricate novel pH-responsive MUP-loaded LPHNs based Hydrogel (MUP-LPHNs-HG) and MUP-LPHNs based film forming spray (MUP-LPHNs-FFS) for enhanced antibacterial activity and effective wound healing. To the best of our knowledge, no scientific investigations have evaluated the use of chitosan and phospholipid-based hybrid systems loaded in hydrogels and film forming sprays for the pH responsive release of MUP against resistant bacterial wound infections. The proposed pH responsive MUP-LPHNs were fabricated and incorporated into Carbopol® 934 Hydrogel and Eudragit S 100 FFS. The developed smart MUP-LPHNs-HG and MUP-LPHNs-FFS showed a smart pH responsive and sustained release of MUP at the wound site, whereas chitosan has positive charge and tends to adhere to the negatively charged bacterial surface. The pH responsive MUP-LPHNs-HG and MUP-LPHNs-FFS can kill bacteria more effectively, prevent bacterial resistance and promote wound healing. NEG is widely used as a tool for topical drug delivery because it can incorporate both lipophilic and hydrophilic drugs. Additionally, it enhances the stability of the formulation through reductions in surface and interfacial tension, which raises its viscosity, eases its application, and avoids gastrointestinal problems. Additionally, several investigations emphasized that NEG provides more stability in formulations than NE, which raises aqueous phase viscosity through a reduction in surface and interfacial tensions. Remarkably, in order to develop pharmaceutical products of the highest quality and with better attributes, it has been discovered that formulation by design (FBD) is a useful technique. This technique helps in the development of more effective, economical, and safe drug delivery systems towards the achievement of quality-by-design goals. This requires a reasonable approach to experimental strategies, such as using a central composite design (CCD) approach.
Mupirocin Liposomes
Compatibility Studies
FT-IR
FT-IR analyses were employed to explore potential interactions between the pharmaceutical and the excipients utilized. The identification of interactions relies on observing changes, shifts, or disform in functional group peaks of the drug. Figure 8 depicts the FT-IR spectra of Mupirocin and its physical mixture with polymers. Noteworthy variations in the position of characteristic peaks for Mupirocin, such as those at 3481 and 3308 cm-1 attributed to the O-H group, peaks at 1729 and 1712 cm- 1 corresponding to the C=O group, and peaks at 1232 and 1222 cm-1 corresponding to the C-O group, were not spot when compared to its physical mixture with the polymers. This indicates the compatibility of Mupirocin with the selected excipients.
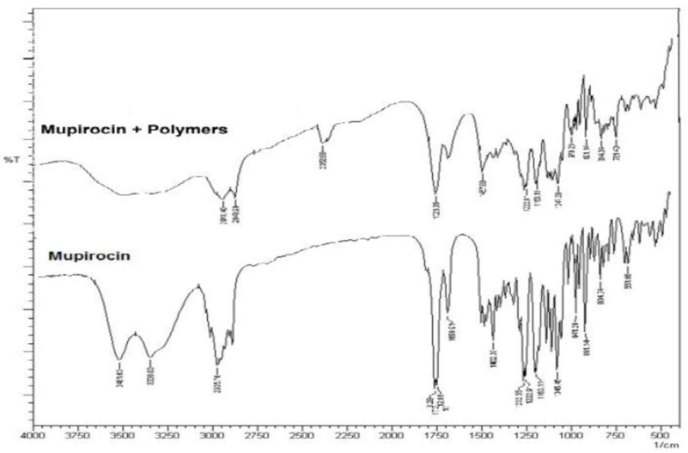
Figure-1: FT-IR spectra of Mupirocin and physical mixture of Mupirocin and polymers.
Contrastingial scanning calorimetry
The DSC thermograms exhibited distinct furthermost partothermic peaks aligning with the Mupirocin melting point at 77.31°C and 75.82°C for its physical mixture with polymers. This suggests an absence of interaction between Mupirocin and the studied polymers. Figure 2 and Table 1 present the DSC thermograms of Mupirocin and its physical mixture with polymers.
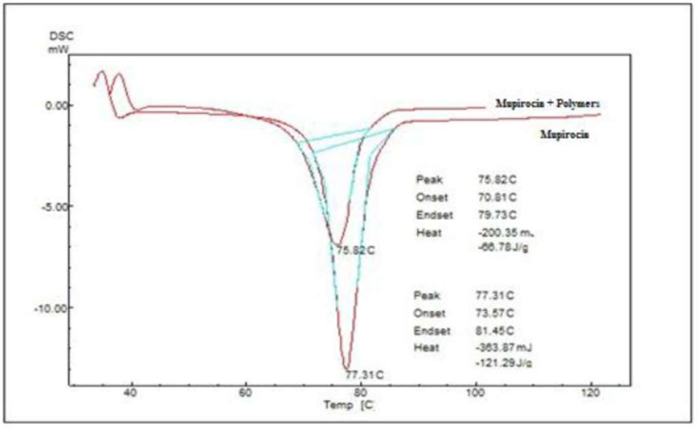
Figure-2: DSC Thermograms of Mupirocin and physical mixture of Mupirocin with polymers.
Table-1: DSC data of Mupirocin and physical mixture of Mupirocin with polymers.
|
SI.NO |
Sample |
To(° C) |
Tm(° C) |
Tc (° C) |
Melting province (°C) |
|
1 |
Mupirocin |
73.57 |
77.31 |
81.45 |
7.88 |
|
2 |
Mupirocin + Polymers |
70.81 |
75.82 |
79.73 |
7.88 |
To - Onset of melt, Tm - Melting point, Tc - Completion of melt, DSC data obtained at 10° C/min
Preparation of Mupirocin liposomes:
A arbitrary 32 full factorial design was executed, with cholesterol represented as A and phospholipon 90H represented as B, selected as unconditional variables, and percent yield (Y1), particle size (Y2), and PDI (Y3) designated as DV. The variables were investigated at three levels (-1, 0, +1), representing low, medium and high, respectively (refer to Table 5). Employing final formulation software, DE 9 (Stat-Ease Inc., Minneapolis, MN, USA), facilitated the execution of statistical final formulation procedures, which encompass RSM, including MRA, ANOVA, and final formulation of statistical data.
Table- 2: Variables Of 32 Factorial Designs of Mupirocin Liposomes
|
Inconditional variable |
|
Levels |
|
|
|
|
Low(mg) |
Medium (mg) |
High(mg) |
|
|
(A) Cholesterol |
200.00 |
300.00 |
400.00 |
|
|
(B) Phospholipon |
400.00 |
500.00 |
600.00 |
|
|
Conditional variable |
|
|
|
|
|
Y1 |
% Yield |
|||
|
Y2 |
Particle Size (nm) Y3 |
|||
|
Y3 |
PDI |
|||
Table-3: Matrix Of 32 FD For Mupirocin Liposomes.
|
|
Factor 1 |
Factor 2 |
|
Run |
(A) Cholesterol mg |
(B) Phospholipon 90H mg |
|
1. |
400 |
500 |
|
2. |
200 |
600 |
|
3. |
300 |
600 |
|
4. |
300 |
500 |
|
5. |
400 |
600 |
|
6. |
200 |
500 |
|
7. |
400 |
400 |
|
8. |
300 |
400 |
|
9. |
200 |
400 |
Experiment was conducted for 9 runs (Table 3) by varying cholesterol and Phospholipon compositions and analyzed for the changed responses.
Experimental design
The outcome data obtained for various responses is represented in Table 4.
Table 4: Spot response in 32 factorial designs for Mupirocin liposomes.
|
|
Factor |
Factor |
Response |
Response |
Response |
|
1 |
2 |
1 |
2 |
3 |
|
|
A: Cholesterol |
B: Phospholipon 90H |
Percent yield |
Particle size |
|
|
|
Std Run |
|
|
|
|
PDI |
|
|
(mg) |
(mg) |
(%) |
(nm) |
|
|
2 1 |
400 |
500 |
70.46 |
364.40 |
0.387 |
|
8 2 |
200 |
600 |
76.67 |
165.51 |
0.210 |
|
5 3 |
300 |
600 |
74.14 |
265.30 |
0.253 |
|
1 4 |
300 |
500 |
71.71 |
237.60 |
0.300 |
|
7 5 |
400 |
600 |
72.19 |
391.41 |
0.370 |
|
4 6 |
200 |
500 |
77.11 |
149.17 |
0.233 |
|
6 7 |
400 |
400 |
68.03 |
314.63 |
0.470 |
|
3 8 |
300 |
400 |
68.28 |
205.11 |
0.371 |
|
9 9 |
200 |
400 |
70.66 |
115.30 |
0.320 |
The outcomes, as illustrated in Table 4, indicates that the selected variables has significant influence on the targeted responses, including % yield, particle size, and PDI datas. These datas provinced from 68.03% to 77.11%, 115.30 to 391.41 nm, and 0.210 to 0.470, respectively. The utilization of the factorial design approach was given in subsequent regression equations.
Percent yield (%) = +65.660 +0.02293 * Cholesterol +0.0267 * Phospholipon 90H
Particle size (nm) = -230.83 +1.067 * Cholesterol +0.3119 * Phospholipon 90H
PDI = +0.365 +7.73333E-004 * Cholesterol -5.466667E-004 * Phospholipon 90H
Negative datas denote an adverse repercussion of specific variable on the response factor, while positive datas signify a + effect of a particular variable. The outcomes of polynomial regression were visually presented through 3-Dimensional graphs and contour plots (depicted in Figures 2 and 3).
ANOVA investigations confirmed the significance (p < 0>
Percent Yield:
The Mupirocin liposomal formulations exhibited a % yield enclosed by the province of 68.03±0.82% to 77.11±0.54% (refer to Table 5). Notably, this province showed significance concerning the polymer ratios utilized. A escalate in the cholesterol ratio correlated with a diminish in percent yield, while a escalate in the phospholipon ratio correlated with a escalate in percent yield.
Particle Size discernment and PDI:
The size of Mupirocin liposomal particle fell enclosed by the province of 116.50 to 391.41 nm. A positive correlation was spot between size of particles and the concentration of the cholesterol and phospholipon. Meanwhile, the PDI of the liposomal formulations provinced from 0.210 to 0.470 (see Table 5). These outcomes suggest that all formulations-maintained homogeneity.
Table-5: Percent yield, particle size and PDI of Mupirocin liposomes.
|
Formulations |
Percent yield* |
Size of particle * |
PDI* |
|
|
(%) |
(nm) |
|
|
LM1 |
70.46±0.56 |
364.40 |
0.387 |
|
LM2 |
76.67±0.77 |
165.51 |
0.210 |
|
LM3 |
74.14±0.61 |
265.30 |
0.253 |
|
|
|
|
|
|
LM4 |
71.71±0.98 |
237.60 |
0.300 |
|
LM5 |
72.19±0.44 |
391.41 |
0.370 |
|
LM6 |
77.11±0.54 |
149.17 |
0.233 |
|
LM7 |
68.03±0.82 |
314.63 |
0.470 |
|
LM8 |
68.28±0.68 |
205.11 |
0.371 |
*SD, (n=3)
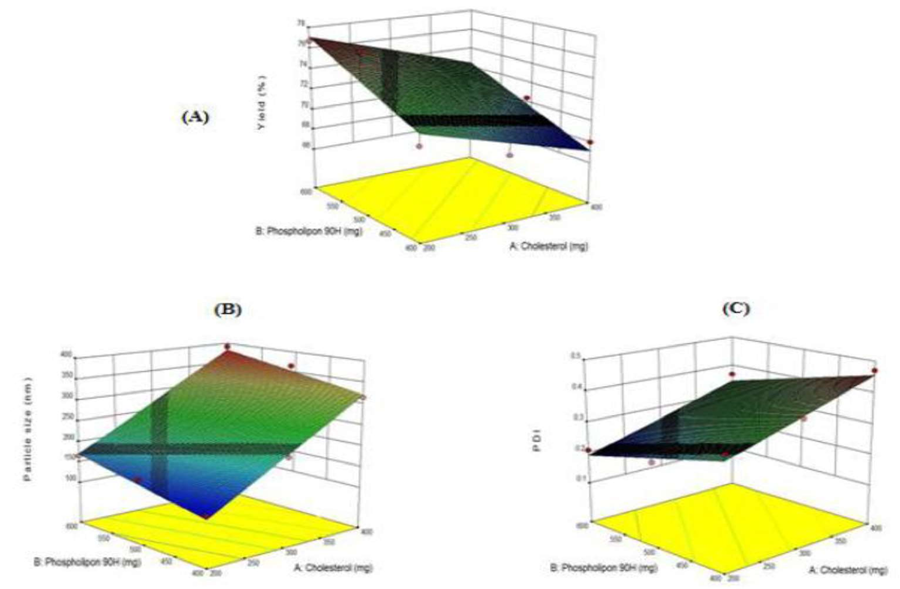
Figure-2: 3-D response surface plot portray the repercussion of cholesterol and phospholipon 90H on (A) percent yield, (B) size of particles and (C) PDI of Mupirocin liposomes.
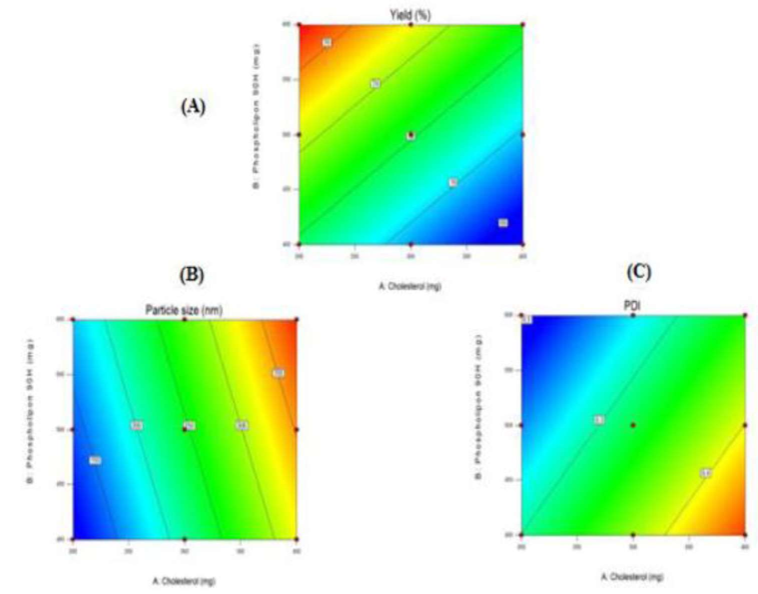
Figure-3: Counterplot representing the repercussion of cholesterol and phospholipon 90H on (A) percent yield, (B) Size of particle size and (C) PDI of Mupirocin liposomes respectively.
The regression analysis disclosed coefficients with a (-) sign for cholesterol and a (+) sign for phospholipon, indicating that the percent yield diminished with an increment in cholesterol concentration, while phospholipon led to an surge in the percent yield. Moreover, an escalate in both cholesterol and phospholipon concentrations emerged in an surge in particle size. However, the rise in phospholipon concentration concurrently surged in the PDI.
Entrapment Efficiency:
The entrapment efficiency of Mupirocin demonstrated variations based on lipid composition, ranging from 52.73±0.66% to 69.14±0.35% (refer to Table no. 6 and Figure no.4). An surge in cholesterol content enclosed by the liposomes correlated with an enhancement in Mupirocin entrapment to certain extent. This effect attributes to the ability of cholesterol to escalate the rigidness of the liposomal membrane, thereby potentially enhancing stead lines and entrapment efficiency. However, over a certain concentration, cholesterol might disrupt the original structure of the liposomal membrane, outcoming in reduced entrapment efficiency.
Conversely, an surge in phospholipon ratio led to higher entrapment efficiency, while an surge in cholesterol ratio, diminishes entrapment efficiency. This observation may be attributed to the saturation of lipids moieties concerning the drug, where a low concentration of phospholipon limits the entrapment capacity.
Contents in drug
The contents in drug in the Mupirocin liposomal formulations was in the scale of 97.23±0.32 - 98.76±0.23% (Table no. 6 and Figure no. 4) representing the Mupirocin was uniformly distributed in the liposomes.
Table-6: Entrapment efficiency and contents in drug Mupirocin liposomes.
|
Formulations |
efficiency* (%) |
Drug content*(%) |
|
LM1 |
55.21±0.52 |
97.61±0.56 |
|
LM2 |
65.85±0.34 |
96.55±0.48 |
|
LM3 |
62.25±0.52 |
98.11±0.13 |
|
LM4 |
59.89±0.34 |
98.36±0.66 |
|
LM5 |
57.46±0.73 |
98.52±0.16 |
|
LM6 |
69.14±0.35 |
97.23±0.32 |
|
LM7 |
52.73±0.66 |
98.14±0.28 |
|
LM8 |
57.53±0.19 |
98.76±0.23 |
|
LM9 |
62.33±0.36 |
97.45±0.33 |
|
*S. D, (n=3) |
|
|
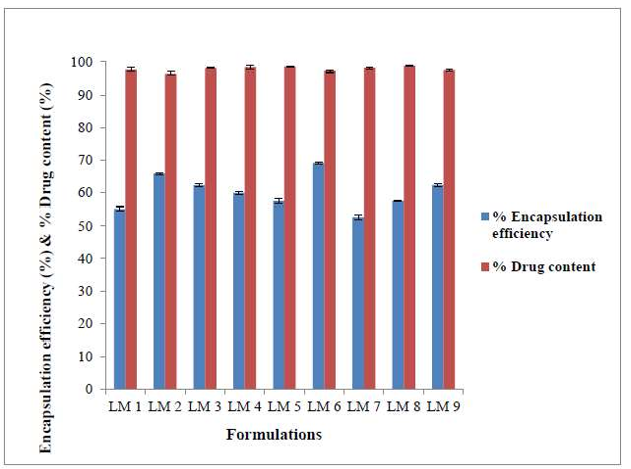
Figure-4: Entrapment efficiency and contents in drug in Mupirocin liposomes. Outline of regression analysis
In the given "F" datas, the associated "p" datas are consistently below 0.05 for all instances, signifying the significance of model terms. The Projected R-Squared demonstrates satisfactory concordance with the changed R-Squared. The assessment of signal-to-noise ratio through Requisite Precision reveals a ratio surpassing 4, which is considered desirable. The obtained value note worthy exceeds 4, providing confirmation of an ample signal (refer to Table 7)
Table-7: Outline of outcomes of regression analysis for responses.
|
|
Value |
F-Value |
P-Value |
|
Percent yield |
|
|
|
|
R-Square |
0.8628 |
|
|
|
Changed R-Squared |
0.8170 |
18.86 |
0.0026 |
|
Envision R-Squared |
0.7039 |
|
|
|
Requisite Precision |
12.248 |
|
|
|
Size of Particle |
|
|
|
|
R-Square |
0.9904 |
|
|
|
Changed R-Squared |
0.9872 |
|
|
|
Envision R-Squared |
0.9789 |
31.37 |
0.0001 |
|
Requisite Precision |
43.704 |
|
|
|
PDI |
|
|
|
|
R-Square |
0.9521 |
|
|
|
Changed R-Squared |
0.9362 |
|
|
|
|
|
59.66 |
0.0001 |
|
Envision R-Squared |
0.8809 |
|
|
|
Requisite Precision |
21.531 |
|
|
Final formulation
To formulate the final dosage forms (liposomes), well-defined limits for the output datas were established (refer to Table no.8). The design expert software was employed to calculate the combinations of variables that led to liposomes meeting the specified criteria. The congruence of the obtained outcomes (see Table no.9 and Figure no. 5) with the envision datas affirms the practicality and ensuring of the model.
Table-8: Final formulation of final Mupirocin liposomes.
|
|
|
Phospholipon |
Percent |
Particle |
|
|
Cholesterol |
|
|
|
PDI |
|
|
Value |
(mg) |
90H (mg) |
Yield (%) |
Size (nm) |
|
|
Envision |
270.85 |
599.99 |
75.45 |
246.48 |
0.247 |
|
Actual |
270.85 |
599.99 |
72.87 |
264.30 |
0.233 |
|
Relative |
- |
- |
2.57 |
17.81 |
0.013 |
|
error (%) |
|
|
|
|
|
Table-9: Particle size with PDI of Mupirocin liposomes.
|
Formulation |
Particle size (nm) |
PDI (PDI) |
|
OLM |
265.50 |
0.263 |
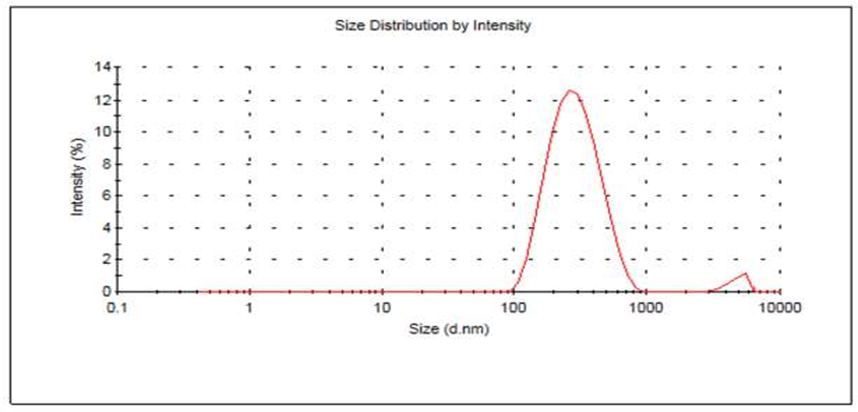
Figure-5: Size Particle distribution of Mupirocin liposomes
In vitro API liberate studies of the final formulation liposome (OLM)
The Mupirocin in vitro dissolution profile from the refined liposomes reached (95.43±0.22%) after 12 hours (refer to Table no. 10 and Figure no. 6). Notably, a escalated ratio of cholesterol in the formulation of the liposomal led to prolonged drug retention, aligning with previous findings that emphasized the role of cholesterol in Ext furthermost parting drug liberate duration99. The delayed liberate of Mupirocin attributes to its lipophilic nature, effectively held by the liposomal membrane's small fragments117. Examining the liberate kinetics, it was evident that Mupirocin liberation from liposomes followed a diffusion-controlled mechanism. A straight correlation emerged when plotting the percent of drug liberated against the square root of time, as per the Higuchi equation (see Table 10). These findings resonate with other studies highlighting the diffusion-controlled liberate mechanism for drugs from liposomes113,118.
Table- 10: In vitro dissolution profile data of refined Mupirocin liposomes (OLM) in phosphate buffer pH 7.4
|
|
CDR* |
|
|
(%) |
||
|
Time (h) |
|
|
|
|
|
OLM |
|
|
0 |
0 |
|
|
1. |
10.65±0.24 |
|
|
2. |
19.89±0.81 |
|
|
3. |
26.75±0.52 |
|
|
4. |
35.59±0.41 |
|
|
5. |
44.85±0.78 |
|
|
6. |
53.78±0.51 |
|
|
7. |
61.97±0.45 |
|
|
8. |
72.84±0.79 |
|
|
9. |
81.75±0.51 |
|
|
10. |
87.59±0.23 |
|
|
11. |
92.84±0.78 |
|
|
12. |
95.43±0.22 |
|
*S. D, n=3 |
|
|

Figure-6: In vitro API liberate profile of final Mupirocin liposomes (OLM) in phosphate buffer pH 7.
Table-11: Liberate kinetic data of final Mupirocin liposomal formulation (OLM)
|
Formulation |
Zero order |
First order |
Higuchi |
Korsemeyer-Peppas |
|
|
|
R2 |
R2 |
R2 |
R2 |
n |
|
OLM |
0.9953 |
0.8455 |
0.9050 |
0.8455 |
1.6355 |
Shape of liposomes:
The shapes of most of the Mupirocin provided liposomes were spherical in shape as shown in Figure 7.
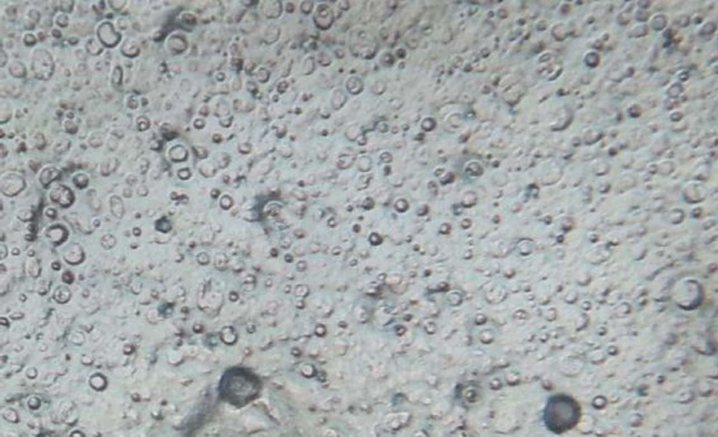
Figure-7: Images of final Mupirocin liposomes (OLM) under 45X 10 magnification
Scanning electron microscopy (SEM)
Liposomes surface morphology and shape were investigated by SEM analysis (Figure 8). The Mupirocin provided liposomes have vesicular structure and were spherical in shape.
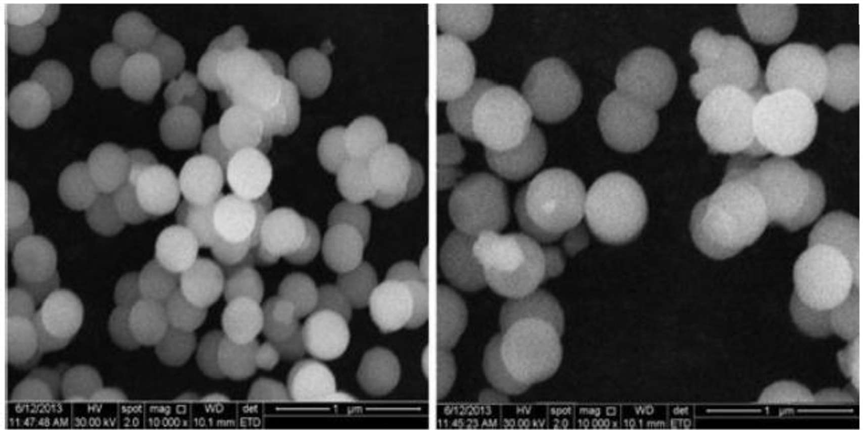
Figure no 8 : SEM images of refined Mupirocin liposomes (OLM)
SUMMARY AND CONCLUSION
The optimized liposomes were spherical in shape and had a vesicular structure. The study demonstrated the importance of optimizing liposomal formulation for improved wound healing outcomes. The results of this study can be used to develop a topical treatment for wound infections, providing a promising approach for the management of this debilitating condition. The use of a 3^2 full factorial design allowed for the evaluation of the effects of cholesterol and phospholipon 90H on the percent yield, particle size, and PDI of the liposomes. The results showed that the selected variables had a significant influence on the targeted responses. The study also investigated the entrapment efficiency and API content of the Mupirocin liposomes. The results showed that the entrapment efficiency ranged from 52.73±0.66% to 69.14±0.35%, and the API content ranged from 97.23±0.32% to 98.76±0.23%. The in vitro release study revealed a diffusion-controlled mechanism, with 95.43% of the drug released within 12 hours. The optimized liposomes were spherical in shape and had a vesicular structure. In conclusion, this study demonstrated the importance of optimizing liposomal formulation for improved wound healing outcomes. The results of this study can be used to develop a topical treatment for wound infections, providing a promising approach for the management of this debilitating condition. The optimized Mupirocin liposomes had a high percent yield, uniform particle size, and low PDI. The entrapment efficiency and API content of the optimized liposomes were also high. The in vitro release study revealed a diffusion-controlled mechanism, with 95.43% of the drug released within 12 hours. The study demonstrated the potential of using liposomes as a topical treatment for wound infections. The results of this study can be used to develop a novel treatment option for wound infections, providing a promising approach for the management of this debilitating condition.
REFERENCES



T. Harika*, Y. Veerendranadh, K. Hema, Aseem Nazia Shaik, D. Ramya, Formulation And Evaluation of Mupirocin Loaded Liposomes by Design Expert Software, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 2, 622-635. https://doi.org/10.5281/zenodo.14842590
 10.5281/zenodo.14842590
10.5281/zenodo.14842590
