Shri Chhatrapati Shahu Maharaj Shikshan Sanstha's Institute of pharmacy, Maregaon.
Neurodegenerative diseases (NDs) such as Alzheimer's disease (AD), Parkinson's disease (PD), Amyotrophic Lateral Sclerosis (ALS), and Huntington's disease (HD) are characterized by progressive neuronal loss, cognitive decline, and motor dysfunction. A growing body of evidence links mitochondrial dysfunction and oxidative stress as common underlying mechanisms in these conditions. Mitochondria, crucial for ATP production and cellular homeostasis, exhibit impaired function and increased reactive oxygen species (ROS) generation in NDs. This review highlights the biochemical and pathological aspects of mitochondrial involvement in NDs and explores the potential neuroprotective role of vitamin E, a fat-soluble antioxidant. Vitamin E, particularly ?-tocopherol, demonstrates the ability to reduce oxidative damage, modulate inflammatory responses, and support neuronal health. Animal model studies further validate the protective effects of vitamin E in mitigating neurodegeneration. However, clinical findings remain inconsistent, emphasizing the need for further research into optimal dosages, timing, and combination therapies. The review concludes with future directions focusing on multi-targeted interventions integrating mitochondrial stabilization, antioxidant therapy, and genetic strategies to manage and potentially slow ND progression.
Neurodegenerative disease:
The progressive death of neurons is a hallmark of neurodegenerative illnesses, which cause behavioural issues, mood changes, cognitive impairments, memory loss, and mobility issues. The main risk factors are environmental variables (toxins), genetic abnormalities in various genes, and, most crucially, aging (Mandemakers et al., 2007). The central nervous system's properties are changed by neurodegeneration, which affects not just the survival or structure of neurons but also their function. Unlike initial cells from the skin, liver, or muscle, neural cells of the central nervous system (CNS) do not multiply after harm from disease, ischemia (deprivation of oxygen, glucose, or blood flow), or physical trauma. Due to the complexity of the human central nervous system, diagnosing and treating neurodegenerative disorders that impair its function has proven challenging: Neurodegenerative disorders cannot be slowed down by any treatment. (Estes & McAllister, 2016).
Neurodegenerative disorders (NDs):
Alzheimer's disease (AD), a neuropsychiatric condition that affects the aged, was first identified in Aloe's Alzheimer in 1911. Silver staining was used in early research on patients with this illness to show that the cerebral cortex had lesions. Neurofibrillary tangles (Nuts), which are histopathologic formations seen inside neuronal cells, were represented by those lesions. An examination of the neuropsychiatric case characterized by Alzheimer's disease using molecular genetics and histology has been published (Grabber et al., 1997). There are several causes and clinical manifestations of Parkinson's disease, which is a recognizable clinical illness. Parkinson's disease is a rapidly expanding neurological disease; aside from an infectious etiology, its increasing global incidence closely mirrors many of the traits usually seen during a pandemic. Numerous observations suggest that Parkinson's disease could not be a unique occurrence. To begin with, parkinsonism is a clinical condition that can seem to have a wide range of causes. There are known causes, such as the less than ten established genes that can definitely cause parkinsonism when they mutate. Second, the disease frequently exhibits a broad range of symptoms and development patterns, even when a cause has been found. (Bloom et al., 2021. The progressive degradation of motor neurons in the brain and spinal cord is a hallmark of amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease in the United States and motor neurone disease in the United Kingdom. ALS is a neurodegenerative illness that progresses and is always fatal. When motor neurons misfire and eventually die, secondary denervation, muscle atrophy, and weakening occur. This typically leads to death from respiratory failure or bulbar dysfunction. (Kim & Taylor, 2017). Huntington's disease (HD) is a crippling neurological condition that affects 10.6–13.7 individuals per 100,000 in Western countries. The mitochondria regulate calcium homeostasis in addition to fuel. demonstrated that lymphoblast mitochondria from HD patients depolarize with lower calcium loading and had a lower membrane potential than control mitochondria. These consist of (I) the build up of lipofuscin, (ii) the presence of the traditional indicator of oxidative modification in lipids (Malondialdehyde and 4hydroxynonenal) and proteins (protein carbonyls), and (iii) signs of oxidative damage to nuclear DNA, like 8hydroxydeoxyguanosine, in HD patients' post-mortem tissue. (Franco-Ibarra et al., 2018).

Fig no.1 “Mitochondrial Dysfunction in Neurodegenerative Diseases.”
General Mitochondrial Biochemistry:
According to one theory, mitochondria were once a single prokaryotic cell that evolved symbiotically with a proto-eukaryotic cell over eons of time. This led to the uptake of the mitochondria and the eventual formation of the first eukaryotic cell. Small molecules and ions can effortlessly pass through the outer membrane of the mitochondria. The presence of an inner membrane that is impermeable to the majority of molecules and ions is the first characteristic that makes the mitochondria special. This membrane, also known as the inner mitochondrial membrane, divides the mitochondria into two main subspaces: the inter mitochondrial space, which is a tiny area between membranes, and the inner space, which is the mitochondrial matrix. The inner mitochondrial membrane contains a number of membrane transport systems, including ATP synthases, ADP-ATP translocases, and the electron transport chain (ETC). The ETC and the ATP synthases dispersed throughout this membrane are essential to this topic because these proteins directly contribute to the utilization of potential energy stored in substrates (like glucose) to drive the synthesis of ATP for biological activity. Glucose is the most significant biologically significant substrate for the mitochondria's bioenergetics activity. Through glucose transport systems, most frequently the GLUT or SLC2A transporter families, glucose molecules are delivered into the cell and then processed catalytically in the cytosol. Upon entering the cell, glucose undergoes catabolism through an enzyme process called glycolysis, which yields two pyruvate molecules, two ATP molecules, two H2O molecules, and two reduced nicotinamide dinucleotide (NADH) molecules. Making NADH, the reduced form of NAD, is a crucial step in connecting the ETC to glucose catabolism (Wattage & Akaka, 2008).
Mitochondrial Dysfunction:
Our cells are powered by mitochondria, which are engaged in the apoptosis-signalling process and produce heat and adenosine triphosphate (ATP) as well as energy The first mitochondrial sickness was identified by Loft and colleagues in 1962 in a 35-year-old female thyroid with myopathy, hyper perspiration, heat intolerance, polydipsia with polyuria, and a basal metabolic rate 180% of normal. The patient had uncoupled oxidative phosphorylation (ox-phis). Ox-phis is the main process that produces cellular energy. A series of respiratory proton (H+) pumps in the mitochondria transports electrons liberated from the reducing substrates, nicotine adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH), to O2 in order to generate ATP, the energy source. (Peacenik & Neustadt, 2007). A wide range of conditions collectively referred to as mitochondrial diseases include oxidative phosphorylation (OXPHOS) abnormalities brought on by mutations in genes encoded by nuclear DNA or mitochondrial DNA (medina). It is unclear what causes cognitive decline in neurodegenerative illnesses as well as in normal aging (Palmer et al., 2021). One clear focal point is a decrease in the quantity and effectiveness of synaptic communication. Genes linked to synaptic plasticity, vesicular transport, and mitochondrial activity were less expressed in the frontal cortex of post-mortem subjects aged 26–106 years after the age of 40. (Keating, 2008).

Fig no 2. “Pathway leading to neurodegeneration in Mitochondria.”
Vitamin E:
Since its discovery about 90 years ago, research on vitamin E has concentrated on a variety of its characteristics, frequently varying according to the particular methods and state of science at the time. In order to provide a concise synopsis and introduction to vitamin E for this special edition of "Molecular Aspects of Medicine" (Zings, 2007). Depending on the particular isoform, vitamin E has a variety of biological actions. Depending on the degree of chemical saturation in their molecular structures, the tocopherol and Tocotrienol subgroups have different characteristics and roles. Tocopherols have phytol side chains, whereas Tocotrienol have three carbon-carbon double bonds (Browne et al., 2019a).
Pathophysiology Of Neurodegenerative Disorder (NDs):
2.1 Alzheimer’s disease (AD):
Alzheimer's disease is a neurological illness that develops and never goes away, affecting many areas of the hippocampus and cerebral cortex. Abnormalities are usually first detected in the frontal and temporal lobes of the brain, and they gradually extend to other areas of the neocortex at rates that vary widely from person to person. Alzheimer's disease is associated with the accumulation of insoluble forms of amyloid-β (Aβ) in blood vessel walls and extracellular spaces, as well as the aggregation of the microtubule protein tau in neurofibrillary tangles in neurons. (Masters et al., 2015). The main source of biological energy production in cells is mitochondria, which is expressed by the coenzyme adenosine triphosphate (ATP). Because biological systems are inherently complex, there are many differences in the characteristics of the mitochondria that are specific to some cell types but absent from others (for example, liver cells are the only cells that exhibit elevated levels of mitochondrial cristae folding). Apart from variations in cell types, there are also changes in mitochondrial structure and/or function brought on by genetic mutations or illnesses like Alzheimer's disease (AD) (Adonic et al., 2016). The hallmarks of AD include aberrant APP metabolism and elevated Ab peptide levels, which are important components of AD neuropathology. Age is the primary risk factor for AD. It is particularly significant that Ab is found in mitochondria and cells. These results, along with the pathogenic part soluble Ab metameric complexes play in the early stages of AD, should provide important hints about the molecular mechanism underlying Ab's neurotoxicity. The specific mitochondrial dysfunctions that have been described are probably compatible with the significant participation in energy hypo metabolism and the increase in free radical production observed in the brain during the early stages of AD. Nevertheless, no overarching mechanism has been put forth to connect Ab to particular important cyst pathologies (such iron homeostasis loss), mitochondrial dysfunction (like complex IV decline), and the neurodegeneration observed in AD. By effectively generating energy, detoxifying oxygen, preserving the cellular redox potential, regulating calcium homeostasis, creating hem and ironsulfur clusters, and other essential metabolites, mitochondria assist their host cells (Ataman & Frey, 2007). By 2050, the United States estimates that the number of new cases will have tripled from approximately 420,000 in 2000 to over 1.3 million. Because women live longer, the incidence of AD is higher in them. It ranges from 1% at years 65 to 70 to about 4% over ages 85. (Imbibe et al., 2005). About 10% of AD diagnoses arise before the age of 65 (early-onset AD), but the bulk of AD cases emerge intermittently after the age of 65. Before receiving a likely AD diagnosis, many AD patients first exhibit subjective memory difficulties. They then gradually advance to moderate cognitive impairment (MCI) at different rates. Slow cognitive decline with deficits in executive function, language, praxis, and visual processing that ultimately result in dementia is the hallmark of Alzheimer's disease, an incurable neurodegenerative illness. (Twohig & Nielsen, 2019).
2.2 Parkinson’s disease (PD):
The causes of Parkinson's disease (PD) are many. Six different gene mutations and five additional chromosome sites have already been linked to familial Parkinson's disease. In terms of pathology, it has been demonstrated that the substantia nigra of patients with sporadic Parkinson's disease displays a variety of biochemical abnormalities, including as mitochondrial dysfunction, free radical damage, inflammatory alterations, and proteasome abnormalities. One noteworthy feature is that the biochemical changes caused by recognized genetic defects substantially align with those previously observed in isolated cases. (Schapira, 2007). The prevalence of Parkinson's disease (PD), the second most common neurological disease after Alzheimer's disease, is 1–2/1000. However, 1–2% of the elderly are impacted because the incidence increases beyond the age of 50. Numerous studies suggest that both inherited and sporadic types of Parkinson's disease may share defects in the ubiquitin-proteasome system (UPS), which breaks down proteins to eliminate excess or misfolded proteins from the brain. (Bartels & Leenders, 2009). Abnormal α-synuclein aggregation, mitochondrial dysfunction, lysosome or vesicle transport issues, synaptic transport issues, and neuroinflammation appear to interact in a complex way to create Parkinson's disease pathogenesis. Together, these disease processes cause dopaminergic neurons to die more quickly, but numerous other motor and non-motor circuits are also affected by the neuropathology. Direct (facilitatory) and indirect (inhibitory) routes across the basal ganglia become unbalanced when nigrostriatal dopamine cells are lost, causing a gradient of striatal dopamine depletion. Bradykinesia was defined by neurophysiological recordings as an imbalance between two oscillatory rhythms: excessive (antikinetic) beta activity and insufficient (prokinetic) gamma activity. Specifically, beta oscillations are associated with the dopaminergic off state and disappear when deep brain stimulation or dopaminergic medication is administered. (Tinkhauser et al., 2017).
2.3 Amyotrophic Lateral Sclerosis (ALS):
Amyotrophic Lateral Sclerosis (ALS), another name for Lou Gehrig's disease, is a neurodegenerative condition that impacts both upper and lower motor neurons. ALS is a multisystemic disease that impacts the cognitive, behavioural, autonomic, and extrapyramidal motor systems, among other areas, according to the Logroscino review. Five to ten percent of all instances of ALS are familial (fALS), which frequently has autosomal dominant genetics. The most common mutations are found in the genes SOD1 (superoxide dismutase-1), C9ORF72 (chromosome 9 open reading frame 72), TARDBP (TAR DNA/RNA-binding protein of 43 k Da), and FUS (RNA-binding protein fused in sarcoma), although there are also reports of very rare or even private mutations in other genes (Jankovic et al., 2021). The cells undergo a retrograde axonal loss, secondary myelin pallor, and gliosis when corticospinal motor neurons deteriorate; these changes occur throughout the spinal cord, but they are most severed at the brainstem and upper spinal cord. Both lower motor neurons (bulb spinal motor neurons) and upper motor neurons (corticospinal motor neurons) degenerate and die in ALS, and reactive gliosis replaces the dead neurons. (H. et al., 2013). Amyotrophic lateral sclerosis (ALS), the most common motor neuron (MN) disease, is considered a rare disease because of its high prevalence (2–3 instances per 100,000 people with European ancestry). The mean age of onset is between 50 and 65, with just 5% of cases occurring at 30 years of age or younger. The majority of the younger patients are men. (Obrador et al., 2020).
2.4 Huntington's disease (HD):
The primary bioenergetics centres regulating the homeostasis of cells and organisms is represented by the double-membrane organelles known as mitochondria. Among other cellular functions, mitochondria regulate the oxidative phosphorylation (OXPHOS) process that produces energy (adenosine triphosphate, or ATP) to support the cells' biosynthetic and degradative metabolic needs, intracellular calcium (Ca2+) homeostasis, apoptosis, and cell signalling pathways. Mitochondrial failure plays a major role in the pathophysiology of Huntington's disease (HD), an autosomal dominant neurodegenerative disorder that initially affects the striatum (mainly the caudate) and eventually the brain. HD is characterized by weight loss, dementia, uncontrollable chloroform movements, cognitive deficits, and psychiatric issues. A polymorphic CAG repeat tract expansion that codes for a Nterminal polyglutamine (poly Q) segment of over 39 residues in the huntingtin protein (HTT) is present in all HD cases, at least in one of their two copies of the HTT gene. This expansion is known as mutant HTT (m HTT) (Carmo et al., 2018). Huntington's disease (HD), a rare autosomal dominant neurodegenerative illness, causes neurons in the striatum and other basal ganglia to gradually degenerate and die. Increased neuronal excitotoxicity, increased oxidative stress, and impaired mitochondrial function are all likely contributing factors to the striatal cell death seen in this specific hereditary disorder (Ansari et al., 2024). A prominent and early reduction in the capacity to recognize emotional emotions in others is one of the devastating, genetic neurodegenerative symptoms of Huntington's disease, which also affects physical, cognitive, and psychiatric functioning. Although we do not yet fully understand the pathophysiology of emotion processing in Huntington's disease, a deeper comprehension of emotion recognition problems will aid in our knowledge of the neurobiology of the condition and the development of treatments to alleviate its symptoms. Asking participants to classify pictures of facial expressions by emotion is the most popular technique for researching emotion recognition in Huntington's disease; both pre-manifest Huntington's disease gene carriers and patients with manifest disease show reduced accuracy (Novak et al., 2012)
Over View Of Vitamin E:
3.1 Introduction:
Soon after Katherine Scott Bishop (1889–1976) and Herbert McLean Evans (1881–1971) recognized vitamin E as a reproductive component in 1922, its antioxidant effects were investigated, particularly by Harold Saft Olcott (1909–1979) and Henry A. Mattill (1883–1953). Initially referred to as "inhibitols," the antioxidant concentration was found to have too similar chemical and physical properties to discriminate between the components of inhibitols from cottonseed, palm oils, wheat germ, and lettuce (Niki & Traber, 2012).
3.2 Chemistry and Structure:
Vitamin E is composed of fats that are slightly soluble in water and soluble in lipoid solvents. Vitamin E components consist of α-, β-, γ-, and δ-homologues, which are bicyclic phenolic compounds grafted to an extended hydrocarbon side chain. These homologues differ from TP and TT in the number and locations of methyl substituents on the chroman ring. While TT has an unsaturated farnesyl side chain, TP has an aliphatic C16 phytyl side chain. The natural α-TP has the following configuration: 2R, 40R, and 80R. As a result, the notation RRR-α-TP, formerly known as D-α-TP, should also be used. Its systematic name is (2R, 40R, 80R)-α-TP. By condensing trimethylhydroquinone with phytol, a combination of racemic TP can be created synthetically. (Khallouki et al., 2020).
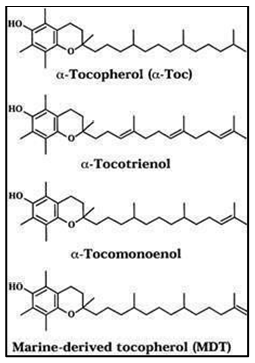
Fig no.3 Structure of vitamin E
3.3 Pharmacokinetics of Vitamin E:
Absorption:
All forms of vitamin E are absorbed by intestinal cells and released into the bloodstream with chylomicrons; at this point, it is likely that there is no discrimination between the different forms. This makes the mechanism of vitamin E absorption shockingly ambiguous. The vitamins enter the liver by chylomicron remnants, where a specific protein called -TTP distinguishes tocopherol from all incoming tocopherols for incorporation into VLDL. Other forms are removed through the bile, urine (as carboxyethyl hydroxyl chromans, or CEHCs), or unknown mechanisms because they are significantly less effectively retained. Furthermore, the plasma has a limited capacity to increase -tocopherol concentrations. Subjects whose typical tocopherol concentration is 25 mol/L cannot have their concentration increased by more than two to three times, regardless of how much or how long they take supplements (Brigelius-Flohé et al., 2002).
Distribution:
Transcription factors known as nuclear retinoic acid receptors alter how they bind to DNA in response to retinoic acid. There are binding proteins for vitamin D as well. These proteins control how vitamin D is transported and distributed throughout cells, and when vitamin D binds, nuclear receptors control gene activity. In this review, a number of proteins that have not been thoroughly described aside from their ability to bind tocopherol are referred to as "tocopherol binding proteins." A family of proteins with molecular definitions that can bind etocopherol is referred to as "tocopherol-associated proteins" (TAPs). Vitamin E is a lipophilic molecule that dissolves poorly in the hydrophilic environment of the cytosol, extracellular fluids, and plasma. It may bind to particular proteins or lipoproteins during absorption, transportation, and distribution, just like other lipophilic vitamins (Behrens et al., 1982).
Metabolism:
It's unexpected. CYP3A is a broad substrate-specific drug-metabolizing system that can be stimulated by steroids, antibiotics such as rifampicin, and other pharmaceutical medications. Whether all rac-tocopherol is just half as effective as RRR-tocopherol is still up for dispute. Given the different ways that specific tocopherols are metabolized, their absorption into plasma lipoproteins, and the possible impacts of their metabolites, it is not surprising when favourable benefits of "vitamin E" are observed in some studies but not in others. (Chiku et al., 1984).
Excretion:
Although EGCG is primarily eliminated through the bile, it can also be eliminated through the urine when taken orally or intravenously. The significant variations in EGCG levels in the feces between oral and intravenous methods imply that the medication taken orally is poorly absorbed (Lambert et al., 2003). The primary biotransformation processes of the ingredients in green tea are methylation and sulfation. I3C is quickly transformed into oxidative metabolites and acid condensation products, however some of the metabolites still have antioxidative qualities, albeit with lower activity than the parent chemical. The metabolism of several polyphenolic glucosides prior to absorption is significantly influenced by the β-glucosidases (Ratnam et al., 2006).
3.4 Mechanism of action of vitamin E:
The liver filters out α-tocopherol and selectively secretes it into the bloodstream inside VLDL and HDL for distribution throughout the body. All eight vitamin E congeners are similarly absorbed from the gastrointestinal tract and transported there by chylomicrons and HDL. Because of this, tissues usually do not contain tocotrienol, while the most prevalent form of vitamin E in human tissues is α-tocopherol, which is followed by γ-tocopherol. The chemical processes driving the preferential retention and cellular sorting of α-tocopherol, one of the eight vitamin E congeners, remain poorly understood even nearly a century after the discovery of vitamin E (Galli et al., 2017).
4. Vitamin E and neurodegenerative disease:

Fig.No.4 “Neuroprotective Role of Vitamin E in neurodegenerative diseases’’.
Mechanisms And Action:
The eight types of vitamin E are fat-soluble compounds that are produced in plants and include α-, β-, γ-, δ-tocopherol, and α-, β-, γ-, δ-Tocotrienol. α-tocopherol, the most common form of vitamin E in human tissues, has long been believed to have cytoprotective properties and may help stop oxidation, inflammation, and degenerative processes. In 1922, Evans and Bishop first identified vitamin E as a dietary component required to stop fetal reabsorption in rats, and it was soon found to be an antioxidant of polyunsaturated oils (Burton, 1994). The substantial health benefits of vitamin E have been shown in numerous studies. First, a hereditary vitamin E deficiency causes a variety of neurological problems. This type of deficiency is brought on by mutations in the TTPA gene, which codes for the tocopherol transfer protein (TTP). The substantial health benefits of vitamin E have been shown in numerous studies. First, a hereditary vitamin E deficiency causes a variety of neurological problems. This type of deficiency is brought on by mutations in the TTPA gene, which codes for the tocopherol transfer protein (TTP). Neurodegenerative diseases, which are typified by the progressive loss of specific neuronal cell types, are associated with protein aggregates. A growing body of evidence suggests that oxidative stress plays a significant role in the etiology of neurodegenerative illnesses (Ricciarelli et al., 2007). Antioxidant vitamins protect cells from oxidative damage by reversing the effects of ROS. Vitamin E is one of the strongest dietary antioxidants that stops proteins, lipids, and nucleic acids from oxidizing because it scavenges oxygen radicals. It has been shown that adding vitamin E to cell membranes protects the cells from damaging oxidation (Brigelius-Flohé, 2006). One essential nutrient that must come from diet is vitamin E. Edible oil from plants, including peanut, sunflower, wheat germ, and safflower oil, is the main source. Butter and cereals come in second and third, respectively. Because vitamin E is fat soluble, its transport and absorption inside the body are highly complicated and controlled by the amount of fat consumed. Micellarization and bile acid secretion are necessary for its absorption in humans, which takes place in the proximal portion of the intestine. Adipose tissue contains the majority of vitamin E, and the Golgi apparatus and lysosomes contain the highest quantity of a-tocopherol among the subcellular membrane fractions (Allan Butterfield et al., 2002).
Alzheimer’s Disease (AD):
The E Vitamins are lipid-soluble, structurally identical antioxidants found in all cellular membranes. They are present in many vegetable oils and are essential for both humans and animals. The eight compounds that make up the vitamin E family are tocotrienol and α-, β-, γ-, and δtocopherols. αtocopherol is the primary E-vitamin present in supplements and the most common form of vitamin E in human tissues. Because of its anti-oxidative qualities, which protect lipids from peroxidation in membranes, vitamin E supplementation has been suggested as beneficial in AD. By influencing several transcriptional pathways, such as the PPARγ (peroxisome proliferator-activated receptor γ) and NF-κB (nuclear factor-κB) pathways, vitamin E molecules exhibit neuroprotective, anti-inflammatory, and hypocholestemic effects in addition to their anti-oxidative qualities. Furthermore, through altering several transcriptional pathways, including the PPARγ (peroxisome proliferator-activated NF-κB (nuclear factor-κB) and receptor γ) pathways, vitamin E can alter gene expression. (Grimm et al., 2016). The primary isoform commonly found in vitamin E supplements is α-tocopherol. The phrase "vitamin E" describes a family of eight naturally occurring homologues with potent antioxidant properties. These include four tocopherols and four tocotrienols, each of which has an α, β, γ, and δ isoform. In food sources such as grains, nuts, seeds, and vegetable oils, all eight congeners are distributed differently. (Browne et al., 2019b). Progressive memory loss and cognitive decline are hallmarks of AD, a neurodegenerative disease linked to aging. Increased oxidative stress and inflammation, which may eventually result in neuronal death, are two of the hypothesized mechanisms behind cognitive decline and AD pathogenesis. Because ROS and peroxide radicals mainly target the polyunsaturated fatty acids of membrane lipids, the brain is particularly vulnerable to oxidative damage. Malondialdehyde concentrations, a gauge of lipid peroxidation, are in fact noticeably higher in AD patients (Boccardi et al., 2016). Pathological characteristics of AD include synapse loss, neurofibrillary tangles, and amyloid b-peptide (Ab) build up in senile (neuritic) plaques. There is strong evidence that AD causes oxidative brain damage, especially Ab, and that this oxidative stress may be linked to Ab's side effects. Ab causes oxidative damage to neurons, rendering them neurotoxic. Vitamin E inhibits these actions in vitro. (Gugliandolo et al., 2017).
Parkinson’s diseases (pd):
There is strong evidence that neurodegeneration in Parkinson's disease is caused by free radical-mediated cell death. Findings such as increased basal concentrations of malondialdehyde and other lipid peroxidation products, decreased levels of antioxidant molecules like glutathione, increased mitochondrial SOD activity, altered iron metabolism, and increased neural phagocyte activity point to a possible role for oxidative stress in Parkinson's disease (PD)-related brain nigral degeneration. Thus, substances with antioxidant qualities, such as vitamin E, may be able to stop Parkinson's disease from happening. (Cadet, 1986). Postural instability, tremor, and bradykinesia are clinical features of Parkinson's disease (PD), a chronic progressive neurological condition. Histopathologically, PD brains show permanent loss of nigrostriatal dopaminergic neurons and intraneuronal accumulation of alpha synuclein proteins. Using both in vitro and in vivo experimental paradigms, studies have demonstrated both vitamin E-mediated protection and lack of protection. In the MPTP-induced PD mouse model (C57/B1), vitamin E deficiency increased MPTP toxicity, as evidenced by mortality and dopamine metabolite depletion in the substantia nigra. Later studies found that pre-treating mice with a daily oral dose of a-tocopherol (48 mg/kg) did not lessen the striatal dopamine depletion caused by MPTP. (Abou-Sleiman et al., 2006). Physiologically, reactive oxygen species (ROS) such superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH) can be produced as a result of the mitochondria's metabolism of molecular oxygen. For example, O2− produced by mitochondrial complexes I and III is converted to H2O2. Furthermore, the conversion of H2O2 to •OH in the presence of reduced metals produces additional highly reactive species (Sutachan et al., 2012). The reduced brain mitochondrial oxidative phosphorylation seen in vitamin E-deficient rats suggests that vitamin E may play a physiological role in sustaining mitochondrial respiration. Long term vitamin E treatment of orga-notypic SN cultures supported this claim by preventing oxidative damage and the death of tyrosine hydroxylase-immunoreactive neurons exposed to rotenone induced chronic complex I inhibition (Camilleri & Vassallo, 2014).
Amyotrophic Lateral Sclerosis (ALS):
ALS is a neurodegenerative disease that affects motor neurons in the cortex, brainstem, and spinal cord. Three to five years following diagnosis, this horrible sickness typically results in death. 20% of instances of familial ALS, which account for 10% of all cases, are associated with a mutation in the SOD1 gene, which codes for CuZn-SOD. The gain of function mutation in CuZn-SOD results in an increase in H2O2. Through Fenton chemistry, H2O2 can produce the reactive hydroxyl radical, which enables indiscriminate attack on surrounding molecules including lipids, proteins, and so forth. Clinical symptoms in patients with sporadic ALS were associated with red blood cell membranes and supernatants that contained protein carbonyls, an indicator of oxidative stress (Abe et al., 1995). Reactive oxygen species (ROS) are radical or non-radical oxygen species produced by the partial reduction of oxygen and are metabolic by products of enzymatic and non-enzymatic activities in cells. These include, for instance, hydroxyl radical (HO•), superoxide radical anion (O 2•-), and hydrogen peroxide (H 2 O2). (Cunha-Oliveira et al., 2020). The progressive loss of upper and lower motor neurons at the bulbar, spinal, and motor cortical levels is a hallmark of amyotrophic lateral sclerosis (ALS), one of the neurodegenerative diseases. The French neurologist Jean-Martin Charcot was the first to describe it, and it is also known as Charcot sickness. Muscle twitching, dysarthria, dysphagia, and regional asymmetric muscle weakness of the upper and lower limbs that advances in myotomal distribution are frequently the initial signs. The disease eventually results in atrophy and impairment of the limb muscles. (Khairoalsindi & Abuzinadah, 2018).
Huntington’s disease (HD):
Clinical signs of HD, a genetic neurodegenerative disease that affects the medium spiny neurons in the striatum, include ataxia, chloroform movements, and dementia. The primary neuropathological features of the disease are astrogliosis, neuronal loss, and significant atrophy in the neostriatum. The genetic anomaly in HD has been shown to be an erroneous extended trinucleotide (CAG) repeat in a gene on the short arm of chromosome 4 that codes for a protein called "huntingtin" (Huntington's Disease). (Huntington's Disease Collaborative Research Group, 1993), whose goal has not yet been established. Several lines of evidence indicate that a failure in mitochondrial energy metabolism may be linked to the pathophysiology of the selective neuronal death observed in HD. N-terminal huntingtin fragments with enlarged polyglutamine tracts can accumulate in the nucleus and cause neuronal death via apoptotic pathways, despite the fact that full-length huntingtin is primarily found in the cytoplasm. Although the exact source of oxidative stress is unknown, it has been linked to the pathophysiology of this illness (Velusamy et al., 2017). Huntington's disease (HD) is another progressive neurodegenerative condition that affects the central nervous system. Similar to PD and AD, HD is characterized by psychiatric problems, dementia, and motor deficits. (Manoharan et al., 2016).
Table no 1. "Comparison of Neurodegenerative Diseases: Affected Brain Regions, Protein Aggregates, and Clinical Manifestations"
|
Disease |
Affected Brain Regions |
Aggregating Proteins |
Type Of Protein Deposits |
Clinical Manifestation |
|
Alzheimer’s Disease [AD] |
Cerebral cortex, Hippocampus
|
Amyloid-ß (A ß) tau
|
Extracellular plaques, Intracellular tangle |
Memory loss and behavioural abnormalities. |
|
Parkinson’s Disease [PD] |
Substantia nigra
|
α -Synuclein |
Intracellular cytoplasmic inclusion (Levy bodies) |
Resting tremor, postural instability, gait disturbance, bradykinesia and rigidity. |
|
Amyotrophic Lateral Sclerosis
|
Spinal cord motor cortex brain stem
|
TDP SOD1, FUS 43
|
Intracellular cytoplasmic inclusion |
Muscle weakness, muscular atrophy, spasticity and eventually paralysis. |
|
Huntington’s Disease [HD] |
Striatum cerebral cortex
|
Huntingtin |
Intercellular nuclear inclusion |
Chorea, psychiatric disturbances and cognitive impairments. |
5. Animal Model Study:
A steady increase in life expectancy over the past few years has led to a large rise in the prevalence of neurodegenerative diseases (NDD). As a result, there are now more health risks and a greater need for research into new and better therapies. Studies have shown that by protecting neurons from oxidative stress-induced damage, vitamin E supplements can help prevent and delay the onset of neurodegenerative diseases. There have been reports of `-tocopherol's (~-Toc) effects on the central nervous system (CNS) for more than 50 years, so the goal of this study is to gather scientific data about how vitamin E supplements affect neuroprotection an neuro degenerative indicators in experimental models. It has been proposed that tocopherol levels and brain health are associated, especially in conditions like ataxias, AD, and PD that are linked to oxidative stress. Consequently, progressive neurological disorders such spinocerebellar ataxia may arise from VE insufficiency since it results in the loss of peripheral nerves. Consequently, long-term Toc administration may halt the progression of degeneration of the VE-deficient nervous system. (da Cunha Germano et al., 2023a). VE supplements can lower oxidative stress and change the central nervous system. AD-induced Tg 2576 mice received a dose of vitamin E (2 IU/g) for eight months. The researchers found a decrease in amyloid plaque deposition, soluble A levels, and lipid peroxidation. VE supplements can lower oxidative stress and change the central nervous system. AD-induced Tg 2576 mice received a dose of vitamin E (2 IU/g) for eight months. The researchers found a decrease in amyloid plaque deposition, soluble A levels, and lipid peroxidation. Additionally, 100 mg/kg/day of -Toc was administered to Wistar rats that had previously experienced oxidative stress and neurotoxicity for ten days. They found that a neuroprotective effect had been observed and that the loss of neurons had diminished after this. (Connolly et al., 2018). VE and -Toc have shown a neuroprotective effect in several neurological models. Vitamin E and -Toc supplements protect against oxidative stress, the main cause of neurodegenerative illnesses, and can reach the entire central nervous system, hippocampus, neurons, memory functions, behaviour, learning, and cognition. VE exhibited a regenerative effect on transgenic mice APPswe/PS1d9 with AD, slowing the disease's course, reducing oxidative stress, and altering neuronal structure to generate neuro protection. (da Cunha Germano et al., 2023b). Another study suggests that in mouse models, VE can restore the effects of traumatic brain damage (TBI) on the molecular substrates that support hippocampal synaptic plasticity and cognitive function. This neurodegenerative effect, which has resulted in a significant improvement in cognitive abilities, a delay in the progression of AD, an increase in cell quantity, a decrease in cell damage, a change in neurite structure, and a decrease in oxidative stress, is supported by other research in this review. They added that low-grade inflammation results in the release of IL-1, IL-6, and TNF-. Conversely, it has been demonstrated that VE and/or LTB supplementation reduces TNF- and IL-6 levels, which in turn reduces DNA fragmentation and MAPK signaling pathways. Thus, VE supplementation prevented oxidative damage to systemic vasculitides in vivo. (Chaturvedi et al., 2010).
Effect of vitamin E on mice model:
|
Species |
Experimental Model |
Dose |
Administration routes |
Outcome measurement &main finding |
|
|
Tg 2576 mice |
|
AD |
2 mg/g |
Oral |
Neuroprotection & Reduced lipid peroxidation soluble A ß and amyloid plaque deposition. |
|
Tg 2576 Mice |
|
AD |
2 mg/g |
Injection into the left pretemporal region |
Neuroprotection & Reduced BLP [Brain Lipid Per oxidant] alleviated the learning deficit improved behavioural commitment. |
|
APPswe/PS1dg mice |
|
AD |
1 day before surgery 210 mg/kg and after for 15 days |
Gavage |
Neurodegeneration & Slowed the progress of AD significantly reduced oxidative stress changed the structure of the neurites |
|
PLTO-KO mice |
AD
|
Memory impairment |
800 mg/kg |
oral |
Neuroprotection & Reduced short term memory impairment prevented PLTP-KO compromise |
Table no 2. "Animal Models in Neurodegenerative Disease Research: Experimental Design, Dosage, and Outcomes"
|
NMRI mice |
PD |
5 mg/kg |
oral |
Signs are antagonized |
|
PINK1-/- mice |
PD |
_ |
chronic |
Restored corticostriatal synaptic plasticity in these mice |
|
Sprague Dawley rats |
PD initial model |
16 mg/kg or 0.8 ml/kg |
intramuscular |
Neuroprotection & Delayed functional decline |
|
Wister rats |
Sintomas semelhantes a PD |
5 and 10 mg/kg |
Intraperitoneal |
Neuroprotection and neuro- inflammation |
|
Albino Swiss mice |
PD |
5,10,20,40 mg/kg |
Oral route |
Neuroprotection& increase the levels of antioxidants enzyme and neurotransmitters decrease the level of inflammatory cytokines. |
|
G93A/SOD1 mouse model |
ALS |
_ |
oral |
Delayed symptom onset and slowed disease progression in this model. |
|
|
|
|
|
Extended survival and slowed disease on set and progression in this model. |
|
SOD1G93A mice |
ALS |
_ |
oral |
|
|
Murine models |
HD |
_ |
_ |
Reduce the volume of striatal lesions |
|
Mouse model |
HD |
_ |
_ |
Impaired glucose uptake and levels |
Future Prospective:
Huntington's disease (HD), Parkinson's disease (PD), Alzheimer's disease (AD), and amyotrophic lateral sclerosis (ALS) are among the neurodegenerative disorders (NDs) that are thoroughly examined in this work. It discusses the molecular mechanisms underlying these disorders, emphasizing mitochondrial malfunction, oxidative stress, and the role of vitamin E in potential therapeutic approaches. Several potential research and therapeutic focus areas are identified in the publication for future consideration: Since mitochondria are essential for cellular health and energy generation, future studies could concentrate on figuring out how mitochondrial dysfunction uniquely affects each neurodegenerative illness. Treatments that stabilize or improve mitochondrial activity might offer a way to decrease the progression of the disease. Further research on antioxidants like vitamin E is relevant since oxidative stress appears to be a major factor in the etiology of many disorders. The document cites studies with conflicting findings, indicating that the secret to efficacy may lie in timing, dose, and combinations with other treatments. Better-targeted antioxidants may be used in future treatments. Vitamin E is a good target for future therapies due to its neuroprotective benefits and function in controlling inflammation. Clinical investigations, however, have produced inconsistent results, highlighting the need for more investigation into the effects of vitamin E types (such α-tocopherol) and dosages on the development of neurodegenerative diseases. Gene Therapy and Genetic Research: Since diseases like ALS and HD are associated with genetic alterations, developments in gene therapy may help develop future treatments by enhancing cellular resilience or targeting certain mutations. Holistic and Multi-Targeted Therapies: Due to the intricacy of many conditions, single-target therapies might not be enough. Multi-targeted treatments that simultaneously address inflammation, oxidative stress, and mitochondrial health may be used in future use.
CONCLUSION:
In conclusion, because of their complexity, neurodegenerative diseases including Huntington's, Parkinson's, ALS, and Alzheimer's are very challenging to diagnose and treat. Similar underlying mechanisms underlie many disorders, including oxidative stress, inflammation, and mitochondrial dysfunction, all of which cause neuronal damage and progressive impairments in motor and cognitive function. Since addressing mitochondrial stability may offer a means of reducing cell death in these disorders, mitochondrial health and bioenergetics continue to be crucial areas for possible intervention. To increase therapeutic effectiveness, future studies should concentrate on multi-targeted approaches that combine metabolic, anti-inflammatory, and genetic techniques. These avenues could lead to more specialized, effective treatments and enhance the quality of life for those with neurodegenerative diseases. Current therapeutic strategies, such as the use of antioxidants like vitamin E, have some promise in preventing oxidative damage, but the inconsistent findings across trials point to the need for more investigation into the best times, dosages, and combinations with other therapies
REFERENCES


Samruddhi Gajbhiye*, Gayatri Jaiswal, Dhanshree Nibrad, Dr. Nilesh Chachda, Exploring Neurodegenerative Diseases: Mitochondrial Dysfunction and The Potential Role of Vitamin E., Int. J. of Pharm. Sci., 2025, Vol 3, Issue 6, 4460-4480. https://doi.org/10.5281/zenodo.15746203
 10.5281/zenodo.15746203
10.5281/zenodo.15746203
