Anuradha College of Pharmacy, Chikhli, Dist. Buldana, MS.
Monoclonal antibodies (MAbs) have emerged as a cornerstone of modern biotherapeutics, offering remarkable specificity and efficacy in the diagnosis and treatment of various diseases, including cancers, autoimmune disorders, and infectious diseases. This review presents a comprehensive overview of mAbs, covering their discovery, structural features, and diverse therapeutic mechanisms such as antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and receptor blockade. The paper discusses key milestones in monoclonal antibody development, from the introduction of hybridoma technology to advanced engineering approaches like phage display, yeast surface display, single B-cell screening, and the use of transgenic animal models. It categorizes mAbs based on their origin—murine, chimeric, humanized, and fully human-as well as their functional types, including unconjugated, conjugated, and bispecific antibodies. Additionally, the review highlights current FDA-approved therapeutic antibodies and their clinical relevance. Emphasis is placed on innovations in production technologies, particularly recombinant DNA techniques, which have enhanced antibody specificity and reduced immunogenicity. With the monoclonal antibody market projected to grow substantially, this review underscores their expanding role in precision medicine and targeted therapies, providing a foundation for future advancements in biomedical science.
Monoclonal antibodies (MAbs) are a group of uniform antibody molecules that specifically bind to a single antigen. They are commonly produced using hybridoma technology, which involves fusing a B-cell with a single lineage of cells that carry a specific antibody gene.[1] MAbs are synthetic proteins that act like the body's natural defences.[2] Monoclonal antibodies are immunoglobulins that are uniquely derived from a monoclonal cell line and provide a broad range of specificity.[3] Because of their precision and consistent production through cell culture methods, mAbs have an advantage over polyclonal antibodies.[1] Monoclonal antibodies (mAbs) are a key breakthrough in modern medicine. They have proven effective in treating a variety of conditions such as cancer, autoimmune diseases, and infections. In many high-income countries, they are now considered the standard treatment for several serious illnesses, offering better results than older therapies—for example, in treating B-cell non-Hodgkin lymphoma or multiple sclerosis. [2]
History of Monoclonal Antibodies
The production of MAbs through hybridoma technology was discovered in 1975 by Georges Köhler of West Germany and César Milstein of Argentina. Together with Niels Kaj Jerne of Denmark, they were awarded the Nobel Prize in Physiology or Medicine in 1984.[1] The first report of laboratory-created mAbs was published in 1970. It described the technique of isolating a single plasma cell clone, which generated a homogenous antibody. This antibody was expanded through repeated passage of spleen cells into irradiated syngeneic mice. However, the first monoclonal antibody intended for human use was not produced until 1975, and it wasn’t fully licensed until 1986. Milstein and Köhler received the Nobel Prize in Physiology or Medicine in 1984 for being the first to successfully produce large quantities of mAbs in vivo. This marked the beginning of the “hybridoma” era and permanently transformed the process of monoclonal antibody production.[4] In 1988, Greg Winter introduced the first humanized MAbs to reduce the immune reactions commonly seen in patients treated with murine-derived MAbs. [1] The first monoclonal antibody approved by the US Food and Drug Administration was muromonab-CD3, used as an anti-rejection drug. The first monoclonal antibody shown to be effective in the treatment of solid tumors was trastuzumab. [4] The first therapeutic mAb, muromonab-CD3 (Ortho clone OKT3), was approved by the US FDA in 1986. [5] Natalizumab was the first mAb approved by the US Food and Drug Administration (FDA) for the treatment of multiple sclerosis (MS) in 2004. This was followed by alemtuzumab in 2014, ocrelizumab in 2017, and most recently ofatumumab in 2021. [6]
Structure of Monoclonal Antibodies
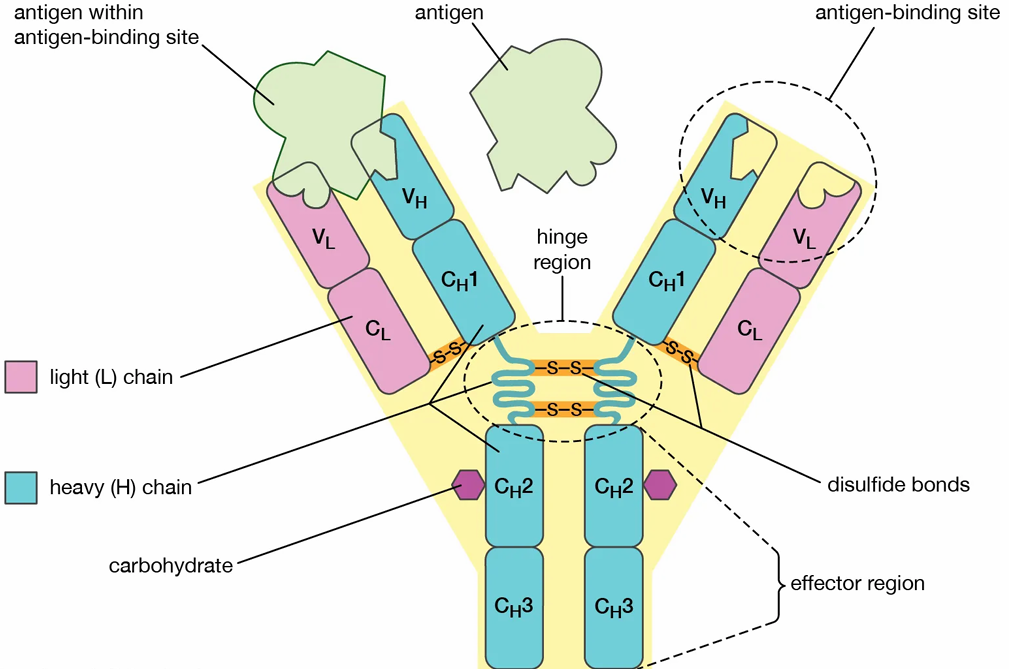
Figure 1: Structure Of Monoclonal Antibody [7]
Monoclonal antibodies have a “Y-shaped” structure made up of four polypeptide chains: two identical heavy chains and two identical light chains. The total molecular weight of a monoclonal antibody is approximately 150 kDa. [8] This structure is entirely protein-based. It cannot be broken down into smaller structural units, but it consists of two different types of chains. These chains are protein in nature and differ in their structural properties. [9] The two arms of the “Y” are known as the Fab (antigen-binding fragment) regions. These regions contain the variable domains that are responsible for antigen binding. The stem of the “Y” is called the Fc (fragment crystallizable) region, which defines the class or isotype of the antibody and carries out effector functions. The Fab region includes complementarity-determining regions (CDRs) that allow the antibody to recognize and bind to a specific antigen with high affinity. The heavy chain forms the lower part of the “Y” and represents the constant region, while the light chain forms the upper arms and contains the variable region that binds to the antigen. [8]
Mechanism of Monoclonal Antibodies
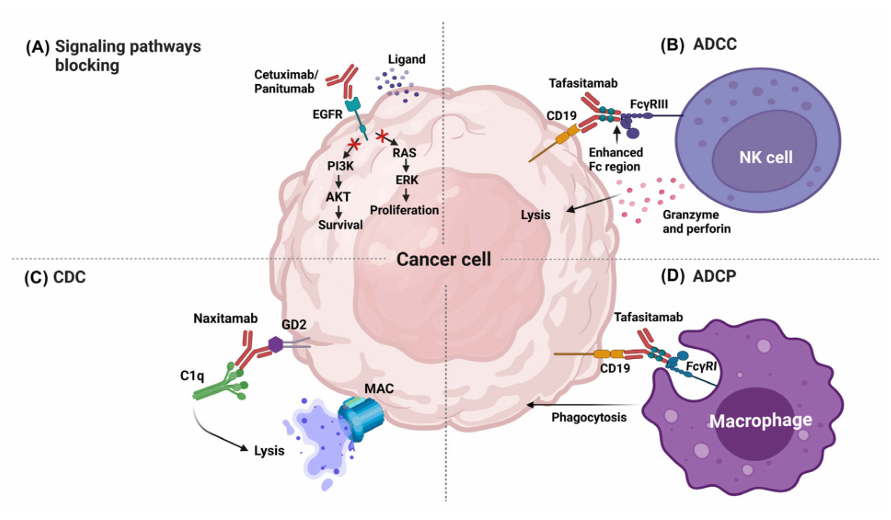
Figure 2: Effector mechanisms of therapeutic mAbs in cancer therapy. [10]
Blocking Signaling Pathways
MAbs can induce the death of tumor cells by blocking the signaling pathways associated with growth factor receptors. Growth signaling and tumor survival could be interrupted when a mAb recognizes, via the Fab region, receptors for growth factors and either inactivates the signaling pathways or blocks the ligand. For example, one of the most commonly targeted receptors using this mechanism is the receptor for epidermal growth factor (EGFR). [11] Some mAbs approved by the FDA act by blocking signaling pathways, such as Cetuximab and Panitumumab. [12]
Antibody-Dependent Cellular Cytotoxicity (ADCC)
Antibody-dependent cellular cytotoxicity (ADCC) is an effector function that results from the antibody binding to both tumor cells and immune cells. The variable regions of the antibody can attach to antigens present on the tumor cell, while the Fc region can bind to Fc receptors (FcR) expressed on leukocytes. For example, Fc RIIIA, expressed in natural killer (NK) cells, triggers cellular destruction by releasing cytolytic factors. [13] Tafasitamab is one of the most recently approved therapeutic mAbs by the FDA. Its target is CD19, a differentiation cluster that is also used as a target in other therapeutic antibodies, such as Loncastuximab and Blinatumomab.[14] Tafasitamab has modifications in the Fc region (two amino acid substitutions: S239D and I332E) that enhance its binding to Fc and improve ADCC. These changes not only increase ADCC activity but also enhance antibody-dependent cellular phagocytosis (ADCP). [15]
Complement-Dependent Cytotoxicity (CDC)
Many therapeutic mAbs used in traditional cancer treatment can also trigger the complement classical pathway (CDC), especially those with the IgG1 isotype. IgG1 antibodies can activate this pathway by binding to Fc receptors in macrophages and NK cells, while also regulating CDC to ensure therapeutic mAbs retain the Fc region of IgG1. The mAbs bind to tumor-associated antigens expressed on the surface of the target cells. Once this binding occurs, C1q can attach to the Fc region of the antibody, initiating a proteolytic cascade. This process facilitates the binding of other complement components until poly-C9 forms a membrane-attack complex (MAC) on the target cell.[16] As an example, Rituximab can work synergistically with ADCC (via NK cells), ADCP (via macrophages), and CDC [17].
Antibody-Dependent Cellular Phagocytosis (ADCP)
ADCP is a biological function that occurs when the Fc region binds to the Fc RI receptor found on macrophages, neutrophils, and eosinophils. ADCP is the process by which antibodies opsonize the tumor cell, allowing it to be internalized and broken down within the phagosome. Generally, it has been noted that antibodies capable of inducing ADCC (such as Tafasitamab) can also enhance ADCP. This effect is linked to the production of gamma-interferon (IFN-γ) by NK cells, which increases the expression of Fc RI in polymorphonuclear cells, thereby promoting phagocytosis. [18]
Classification and Types Monoclonal Antibodies
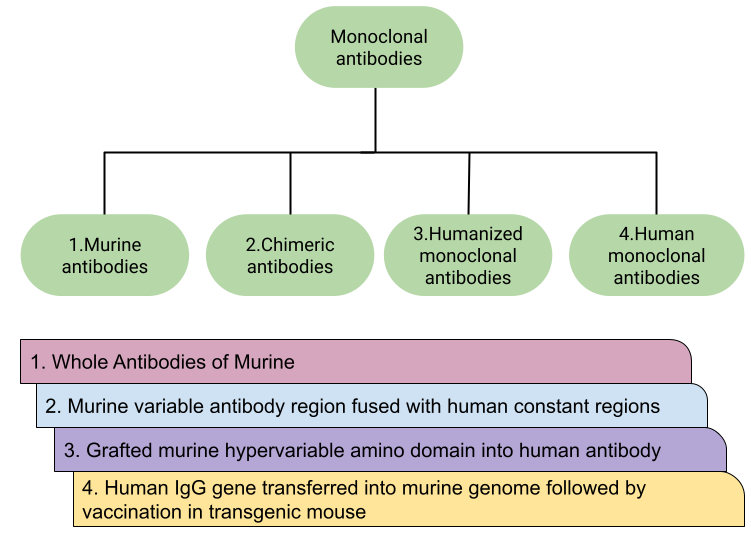
Figure 3: Types of therapeutic MABS and their production methods.[19]
The first mAb to be discovered and developed was the murine monoclonal antibody. This type of mAb is produced by collecting B lymphocytes from the spleen of a mouse and fusing them with an immortal myeloma cell line that lacks the hypoxanthine-guanine-phosphoribosyltransferase (HPTR) gene. All murine mAbs are named with the suffix -omab (e.g., muromonab-CD3, blinatumomab, capromab). The variable regions of both the heavy and light chains are derived from mice. Murine mAbs also have a short half-life when used in humans because they bind relatively weakly to the human FcRn. In oncology, these mAbs are often less useful because they are relatively poor at recruiting effector functions such as antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity—both essential for tumor cell destruction. [4]
Chimeric mAbs contain human constant regions and mouse variable regions. This is achieved through genetic engineering techniques, resulting in mAbs that are approximately 65% human and 35% mouse. Chimeric mAbs are typically named with the suffix -ximab (e.g., rituximab, infliximab, cetuximab). Compared to murine mAbs, chimeric mAbs have a longer half-life in humans and reduced immunogenicity. However, the risk of triggering anti-drug antibodies remains significant. [20, 4]
Humanized mAbs are created by grafting the hypervariable regions of mouse antibodies (both heavy and light chains) onto a human antibody framework, making them approximately 90–95% human. These mAbs are named using the suffix -zumab (e.g., trastuzumab, alemtuzumab, bevacizumab). This approach helps reduce immunogenicity and improves therapeutic effectiveness.
These mAbs are generated using human antibody genes, including parts of the variable regions, allowing for proper recombination of human antibodies. Human mAbs are less likely to trigger immune responses and are generally better tolerated than other mAb classes. They are identified by names ending in -umab (e.g., ofatumumab, daratumumab, denosumab). There are three main types of mAbs based on how they are administered or used: unconjugated (or naked), conjugated, and bispecific.
Unconjugated mAbs, or “naked” mAbs, are antibodies that act on their own without being linked to other substances. These are the most widely used type in cancer treatment.
When an mAb is combined with a chemotherapy agent or a radioactive particle, it is called a conjugated monoclonal antibody. The mAb acts as a carrier for the chemotherapy or radioactive substance, traveling through the patient’s body until it reaches the specific target antigen. This approach helps reduce damage to healthy cells by ensuring the chemotherapy or radioactive agent is delivered directly to the intended site via the mAb.
Bispecific mAbs are specially designed molecules that can bind to two different epitopes at the same time. This concept was first described decades ago by Nisonoff et al. [21] One of the targets is a protein found on cancer cells, and the other is a protein found on immune cells. This dual targeting allows cancer cells and immune cells to come into close contact, which can trigger a stronger immune response and lead to more effective destruction of cancer cells. [4]
Production Methods of Monoclonal Antibodies
The following sections outline various MABS production technologies
Figure 4: Production methods MAbs. [22]
Hybridoma technology is a fundamental method for the production of monoclonal antibodies. This technique was first introduced by Köhler and Milstein in 1975. A mouse is carefully immunized with specific target antigens, leading to the development of an immune response. B cells are then collected from the mouse and fused with immortalized myeloma cells that lack hypoxanthine-guanine phosphoribosyltransferase (HGPRT), making them unable to survive in selective culture media. The fusion process is aided by polyethylene glycol, resulting in the formation of hybridoma cells, which are capable of producing monoclonal antibodies. [3]
Immunization of Mouse
A mouse (commonly the BALB/c strain) is immunized with a specific antigen to stimulate B cells to produce antibodies against it. Several booster injections are given to strengthen the immune response.
Isolation of Spleen Cells
After adequate immunization, the mouse is sacrificed and the spleen (a rich source of B cells) is removed. The spleen cells, which include antibody-producing B lymphocytes, are then isolated.
Fusion with Myeloma Cells
The spleen B cells are fused with immortal myeloma cells (cancerous plasma cells that lack the ability to produce antibodies) using PEG (polyethylene glycol). This fusion forms hybridoma cells, which are immortal like myeloma cells and can produce specific antibodies like B cells.
Selection of Hybridomas
The fused cells are grown in HAT medium (Hypoxanthine-Aminopterin-Thymidine), which allows only successfully fused hybridoma cells to survive. Unfused myeloma cells die because they lack the HGPRT enzyme; unfused B cells naturally die as they are not immortal.
Screening and Selection
Hybridoma clones are tested (using ELISA or similar methods) to find those that produce the desired antibody. Clones that test positive are selected and allowed to multiply.
Cloning of Positive Hybridomas
Selected hybridomas are cloned (e.g., by limiting dilution) to ensure monoclonality each clone produces a single type of antibody.
Production and Purification of Monoclonal Antibodies
Clones are grown in large volumes or injected into the peritoneal cavity of mice to produce ascitic fluid, which is rich in antibodies. The antibodies are then purified using methods such as affinity chromatography.
Characterization and Storage
Monoclonal antibodies are examined for specificity, affinity, and isotype. Hybridoma cells are cryopreserved for future antibody production. [23, 24]
Recombinant DNA technology involves the complex process of combining DNA fragments from different sources, including distinct species, to create new genetic sequences that do not occur naturally. This method consists of a series of carefully coordinated steps: isolating the desired DNA, cutting it precisely at specific recognition sites using restriction enzymes, amplifying gene copies through polymerase chain reaction (PCR), joining DNA fragments into a vector, and then inserting the recombinant DNA into a host organism. Such capabilities have wide-ranging applications across various fields, including medicine, agriculture, and industrial biotechnology. Significant uses of recombinant DNA technology include the production of therapeutic proteins (e.g., insulin, growth hormones), gene therapy, genetic modification of crops, and the development of diagnostic tools like ELISA.The ability to manipulate DNA sequences has brought about a major shift in our understanding of genetics and has driven substantial progress in molecular biology, genomics, and proteomics. [8]
The immortalization of B lymphocytes has generally been achieved either through somatic cell fusion with a myeloma, LCL, or heteromyeloma cell line using PEG as the chemical fusogen, or through infection with the B-tropic EBV. While both methods have their own advantages and limitations (in comparison to the relatively simple murine system), the somatic cell fusion method is typically preferred. Additionally, B cells can also be immortalized using ECF technology. [25]
YSD technology is a highly effective tool used in molecular biology and protein engineering. It enables the presentation of proteins or peptides on the surface of yeast cells, allowing for the selection and isolation of desired variants using high-throughput screening methods. This technology has transformed several fields, including antibody engineering, vaccine development, enzyme optimization, and directed evolution. The basic concept of YSD is fairly straightforward. It involves genetically linking the gene encoding the protein of interest with a gene encoding a yeast cell wall protein. This fusion gene is then expressed in yeast cells, leading to the display of the protein on the cell surface. The most widely used yeast species for this application is Saccharomyces cerevisiae, a well-studied and easily modifiable organism. The process of YSD involves several steps. First, the gene encoding the protein of interest is fused with a gene encoding a yeast cell wall protein, typically Aga2p or Flo1p. This fusion gene is then introduced into yeast cells using various methods such as electroporation or lithium acetate transformation. [26]
Phage display, introduced by George Smith in 1985, led to the development of antibody phage display libraries. It offers an alternative approach to traditional hybridoma technology. This method enables the isolation of fully human-derived monoclonal antibodies (mAbs) from large Ig gene repertoires presented on the surface of bacteriophages. [27] Phage display is generally used as a novel strategy to present peptides (polypeptides) on the surface of bacteriophages. In this system, phages can display the desired (poly)peptides as part of their surface proteins. In this technique, the (poly)peptide is fused with one of the surface proteins of the bacteriophage, allowing the peptide to be displayed on the phage surface. In this way, the desired peptide is selected based on its ability to bind to a specific target. In fact, this method is based on the interaction between the ligand and the receptor. Displaying proteins on the surface of phages is a reliable approach for selecting rare traits that encode proteins with binding activity. Phage display can be adjusted to present various peptides with significant differences in polyvalent configurations. The extent of display depends on the sequence and the length of the poly(peptides). Antibody phage display is also gaining growing interest in the field of toxinology. [28,29]
Mammalian cell display antibody library technology involves introducing the target antibody gene into an expression vector and delivering it into mammalian cells (such as CHO or HEK293) through transfection or viral infection. This enables the surface expression of exogenous proteins, thereby creating display libraries. Due to its native eukaryotic secretion system, mammalian cell display technology helps overcome issues like low expression efficiency and protein misfolding, which can occur due to the lack of post-translational modifications in phage display systems. In theory, complex and highly stable antibodies (including Fab, scFv, and full-length IgG antibodies) can be displayed directly on the cell surface using mammalian cell surface display systems. [30]
The phage display system is widely used for generating recombinant monoclonal antibodies and antigen-binding antibody fragments. However, alternative display methods, such as bacterial cell surface display, have also been developed and may offer specific advantages over phage display. Bacterial cell surface display takes advantage of the bacteria’s natural surface-displaying proteins. The core principle is that the gene encoding the desired protein is inserted into the bacterial cell, where it is translated and anchored to a naturally occurring surface protein. Different bacterial display hosts are chosen based on their compatibility with the protein of interest and their proteolytic activity. [31]
Ribosome display was developed to eliminate the need for the transformation step by creating antibody–ribosome–mRNA (ARM) complexes for selection in a cell-free system. ARM complex formation occurs by stalling a ribosome at the end of translation, which links each antibody fragment to its corresponding mRNA. This allows for the simultaneous isolation of both the functional antibody and its encoding mRNA through affinity capture using a ligand. The ribosome-bound mRNA is then converted into DNA via reverse transcription (RT-PCR). Like other display methods, this process can be repeated to enrich for high-affinity antibodies. Both prokaryotic and eukaryotic ribosome display systems have been developed and are now widely used for in vitro selection and the directed evolution of proteins, including antibodies. [32]
The DNA-encoded mAb approach delivers genetic constructs that express the desired mAbs within host cells, eliminating the need for traditional manufacturing and purification processes. Viral vectors, especially adeno-associated virus (AAV), have been successfully used for mAb delivery. [33]
Transgenic animal are one of the fastest growing method in the biotechnology areas. It is used to incorporate exogenous genes into the animal genome by genetic engineering technology so that these genes can be inherited and expressed by offspring. In this approach, human Ig genes are inserted into the animal’s genome to replace the native Ig genes, enabling the animal to produce fully human mAbs upon immunization. The process of producing monoclonal antibodies using transgenic mice is similar to the mouse hybridoma technique. The main use of fully human mAbs derived from transgenic animals is in therapeutic applications. [34]
Single B-cell antibody generation technology is an advanced approach that offers several benefits, including rapid production, high efficiency, and high yield. Antibodies produced using this method maintain their natural conformation and are well suited for applications in pathogen detection, disease treatment, and studies on virus cross-species transmission mechanisms. [35] Single B-cell screening has become an appealing alternative to hybridoma technology for analyzing B cell receptor (BCR) repertoires and identifying potential therapeutic mAb candidates. In a typical process, B cells are isolated using magnetic-activated cell sorting (MACS), labeled with both antigen and antibodies for positive and negative selection based on cell surface markers, and sorted one cell per well using fluorescence-activated cell sorting (FACS). [36]
Monoclonal Antibodies Approved By FDA
Table 1: Monoclonal antibodies approved by the FDA
|
mAb Name |
Target/Epitope |
Antibody Kind |
Cancer Kind |
Year Approval |
|
Alemtuzumab |
CD52/C-terminal with part of the GPI anchor [37] |
Humanized IgG1 |
Chronic myeloid leukemia |
2001 |
|
Atezolizumab |
PD-L1 3/Beta-sheet C` and B-C loop [38] |
Humanized IgG1 |
Bladder |
2016 |
|
Avelumab |
PD-L1/Central beta-sheets C and F [38] |
Human IgG1 |
Merkel cell carcinoma |
2017 |
|
Amivantamab-vmjw |
EGFR/Residues K443, K465, I467, S468 [39] and MET |
Human Ig G1-based bispecific antibody |
Metastatic non-small cell lung |
2021 |
|
Bevacizumab |
VEGF-A/Hairpin loop (β5–turn–β6) and β2–α2–β3 [40] |
Humanized IgG1 |
Colorectal |
2004 |
|
Blinatumomab |
CD19, CD3/Residues 97–107, 155–166, and 216–224 [41] |
Murine bispecific tandem scFv |
Acute lymphoblastic leukemia |
2014 |
|
Belantamab mafodotin-blmf |
BCMA4 [42] |
Afucosylated IgG1 |
Multiple myeloma |
2020 |
|
Brentuximab vedotin |
CD30/Extracellular domain [43] |
Chimeric IgG1 |
Hodgkin lymphoma, systemic anaplastic large cell lymphoma |
2011 |
|
Cetuximab |
EGFR/Domain III amino acids 334–504 [44] |
Chimeric IgG1 |
Colorectal |
2004 |
|
Cemiplimab |
PD-1/BC and FG loops (N58 Glycan) [45] |
Human mAb |
Cutaneous squamous cell carcinoma |
2018 |
|
Dinutuximab |
GD2 [46] |
Chimeric IgG1 |
Neuroblastoma |
2015 |
|
Daratumumab |
CD38/C-terminal loop (residues 189–202 and 223–236) [47] |
Human IgG1 |
Multiple myeloma |
2015 |
|
Durvalumab |
PD-L1/Central beta-sheets C and F [38] |
Human IgG1 |
Bladder |
2017 |
|
Dostarlimab-gxly |
PD-1/PD-L1/BC, C`D and FG loops [48] |
IgG4 humanized |
Advanced solid tumors |
2021 |
|
Elotuzumab |
SLAMF7/IgC2 domain [49] |
Humanized IgG1 |
Multiple myeloma |
2015 |
|
Enfortumab vedotin-ejfv |
Nectin-4/V-domain [50] |
Human IgG1 |
Cancers expressing Nectin-4 |
2019 |
|
Gemtuzumab ozogamicin |
CD33/Ig-like V-set domain [51] |
Humanized IgG4 |
Acute myeloid leukemia |
2017 |
|
Ibritumomab tiuxetan |
CD20/Same as Rituximab [52] |
Murine IgG1 |
Non-Hodgkin lymphoma |
2002 |
|
Ipilimumab |
CTLA-4/front β-sheet [53] |
Human IgG1 |
Metastatic melanoma |
2011 |
|
Isatuximab-irfc |
CD38/C-terminal loop (residues 81–90) [47] |
Chimeric IgG1 |
Multiple myeloma |
2020 |
|
Inotuzumab ozogamicin |
CD22/V-like domain [54] |
Humanized IgG4 |
Acute lymphoblastic leukemia |
2017 |
|
Loncastuximab tesirine-lpyl |
CD19/RB4 [55] |
Humanized IgG1 |
Large B-cell lymphoma |
2021 |
|
Margetuximab-cmkb |
HER2/Extracellular domain [56] |
Chimeric Fc-engineered IgG1 |
Metastatic HER2-positive breast |
2020 |
|
Mirvetuximab soravtansine-gynx |
FRα 5 [57] |
IgG1, Antibody-drug conjugate |
Epithelial ovarian, fallopian tube, or peritoneal |
2022 |
|
Mosunetuzumab-axgb |
CD20/CD3 [58] |
Bispecific CD20-directed CD3 T-cell engager |
Relapsed or refractory follicular lymphoma |
2022 |
|
Nivolumab |
PD-1/BC-loop [59] |
Human IgG4 |
Melanoma, non-small cell lung |
2014 |
|
Necitumumab |
EGFR/Domain III [60] |
Human IgG1 |
Non-small cell lung cancer |
2015 |
|
Naxitamab |
GD2 [61] |
Recombinant humanized IgG1 |
Neuroblastoma |
2020 |
|
Ofatumumab |
CD20/FLKMESLNFIRAHT region [62] |
Human IgG1 |
Chronic lymphocytic leukemia |
2009 |
|
Obinutuzumab |
CD20/Large extracellular loop (172–176 region) [52] |
Humanized IgG1 Glycoengineer-ed |
Chronic lymphocytic leukemia |
2013 |
|
Olaratumab |
PDGFRα 2/Extracellular domain [63] |
Human IgG1 |
Soft tissue sarcoma |
2016 |
|
Panitumumab |
EGFR/Domain III, P349, P362, D355, F412 and I438 [64] |
Human IgG2 |
Colorectal |
2006 |
|
Pertuzumab |
HER2/Extracellular domain II [65] |
Humanized IgG1 |
Breast |
2012 |
|
Pembrolizumab |
PD-1/C, C`, and G antiparallel beta sheets and C-C` and F-G loops [38] |
Humanized IgG4 |
Melanoma |
2014 |
|
Polatuzumab vedotin-piiq |
CD79β/ARSEDRYRNPKGS [66] |
Humanized IgG1 |
Diffuse large B-cell lymphoma |
2019 |
|
Rituximab |
CD20/169-PANPSE-174 and 183-CYSIQ-187 [67] |
Chimeric IgG1 |
Non-Hodgkin lymphoma |
1997 |
|
Ramucirumab |
VEGFR2/Domain III [68] |
Human IgG1 |
Gastric |
2014 |
|
Sacituzumab govitecan |
Trop-2/Q237-Q252 [69] |
Humanized IgG1 |
Solid tumors |
2020 |
|
Trastuzumab |
HER2 1/extracellular domain [56] |
Humanized IgG1 |
Breast |
1998 |
|
Tafasitamab-cxix |
CD19 [70] |
Fc-modified IgG1 |
Diffuse large B-cell lymphoma |
2020 |
|
Tisotumab vedotin-tftv |
Tissue Factor [71] |
IgG1 |
Cervical |
2021 |
|
Teclistamab-cqyv |
BCMA [72] |
Humanized Ig G4-proline, alanine, alanine |
Multiple myeloma |
2022 |
1Human Epidermal Growth Factor Receptor 2; 2 Programmed cell death ligand; 3 Platelet-derived growth factor receptor alpha; 4 B-cell maturation antigen; 5 Folate receptor-alpha.
Table 2: Therapeutic Monoclonal Antibodies withdrawn or discontinued from marketing in the European Union or United states. [73]
|
International proprietary name (Trade name) |
Manufacturing cell line |
Types |
Target |
First EU (US) approval year |
|
Muromonab-CD3 (Orthoclone OKTsg |
Hybridoma |
Murine IgG2a |
CD3 |
1986(1986) |
|
Edrecolomab |
Hybridoma |
Murine IgG2a |
EpCAM |
1995(NA) |
|
Efalizumab (Raptiva') |
CHO |
Humanized IgG1k |
CD11a |
2004(2003) |
|
Gemtuzumab ozogamicin (Mylotarg') |
NS0 |
Humanized IgG4k |
CD33 |
NA (2000) |
|
Nebacumab (Centoxin) |
Hybridoma |
Human IgM |
Endotoxin |
1991(NA) |
|
Daclizumab (Zenapax) |
NS0 |
Humanized IgG1k |
IL2R |
1999(1997) |
Abbreviations: CD, dluster of differentiation; CHO, Chinese hamster ovary; EpCAM, epithelial cell adhesion molecule; IL, interleukin; NA, not approved.
Therapeutic Applications of Monoclonal Antibodies
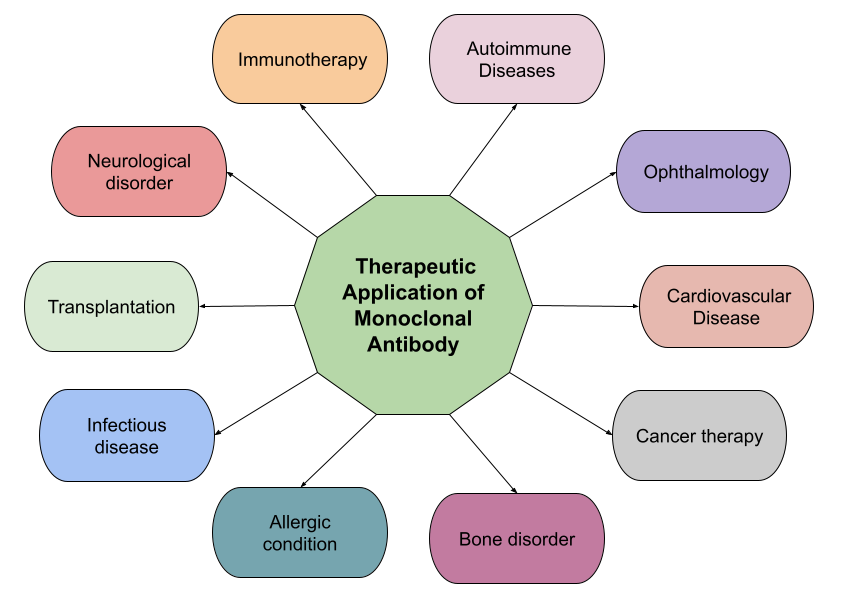
Figure 5: Shows therapeutic applications of monoclonal antibodies. [8]
Future perspectives
Monoclonal antibodies offer significant potential for strengthening defenses against biological warfare agents and emerging infectious diseases. Remarkable progress in antibody engineering has produced more human-like mAbs with reduced immunogenicity, including chimeric, humanized, and fully human antibodies . These next-generation mAbs are being developed for a broad range of uses, from cancer to autoimmune and infectious diseases. The expected rise of the global therapeutic monoclonal antibody market to $300 billion by 2028 highlights the rapid growth and strong potential of this groundbreaking therapeutic approach. Ongoing advances in antibody engineering, combined with the integration of genomic and medical data. [74,75]
REFERENCES



B. P. Barote*, I. H. Bhopale, Aijaz A. Sheikh, K. R. Biyani, Exploring Monoclonal Antibodies Through Literary and Scientific Texts, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 3532-3549. https://doi.org/10.5281/zenodo.16440469
 10.5281/zenodo.16440469
10.5281/zenodo.16440469
