SGMSPM’s Dnyanvilas College of Pharmacy, Pune, Maharashtra, India.
This study aimed to design and evaluate novel antitubercular agents using computational methods. Four derivatives of isoniazid and acyclovir were designed and subjected to molecular docking studies against the 3IX2 protein. The results showed that the first derivative had the lowest binding energy (-11.58) and formed three hydrogen bonds with the protein. ADMET analysis was performed to evaluate the pharmacokinetic properties of the designed compounds. Based on the molecular docking and ADMET analysis, the first derivative was identified as the most promising compound for further development as an antitubercular agent. The study suggests that this derivative could be a potential lead compound for the treatment of tuberculosis.
Tuberculosis (TB) is a chronic infectious disease caused by bacteria from the Mycobacterium tuberculi complex. The primary causative agent, Mycobacterium tuberculi, is a slow-growing, aerobic, non-motile bacillus that is acid-fast due to its high mycolic acid content. Discovered by Robert Koch in 1882, TB is transmitted person-to-person via aerosol droplets. While M. tuberculosis is the main culprit, other species in the complex, such as M. bovis, M. africanum, M. microti, M. Canetti, M. caprae, and M. pinsnipedii, can also cause TB in humans and animals. TB primarily targets the lungs (pulmonary TB) but can also affect other parts of the body like the brain, kidneys, spine, lymph nodes, and bones (extrapulmonary TB). The pathogenesis of TB begins with inhalation of bacilli from an infected person, which reach the alveoli and are engulfed by macrophages. Instead of being destroyed, the bacteria multiply inside macrophages, and the immune system responds by forming granulomas to contain the infection. TB can be latent, with no symptoms and no contagiousness, or active, with symptoms and contagiousness. Diagnosing TB involves various tests, including the Tuberculin Skin Test (TST), Interferon-Gamma Release Assays (IGRAs), sputum smear microscopy, sputum culture, chest X-ray, and molecular tests like GeneXpert MTB/RIF. Treatment typically follows a standard 6-month regimen, consisting of an intensive phase with isoniazid, rifampicin, pyrazinamide, and ethambutol, followed by a continuation phase with isoniazid and rifampicin. However, drug-resistant TB requires longer and more complex treatment with second-line drugs. TB can weaken the immune system, making individuals more susceptible to other opportunistic infections like cytomegalovirus (CMV) , Cancer , Candidiasis , Hepatitis , Herpes Simplex virus ( HSV ) , Herpes zoster (Shingles) Human Papilloma Virus (HPV), Kaposi Sarcoma , Mycobacterium Avium Complex , Non-Hodgkin Lymphoma (NHL). Despite being preventable and curable, TB remains one of the top infectious disease killers globally. According to the World Health Organization's 2023 reports, approximately 10.6 million people fell ill with TB in 2022, and 1.3 million deaths occurred among HIV-negative individuals. Access to timely diagnosis and treatment remains a significant challenge in many parts of the world.
2. MATERIAL AND METHODS:
2.1 Design of Ligand:
Here isoniazid and acyclovir introduce as a parent ring in compounds and both are joined via different linkers showing possible antitubercular activity against Mycobacterium tuberculi.
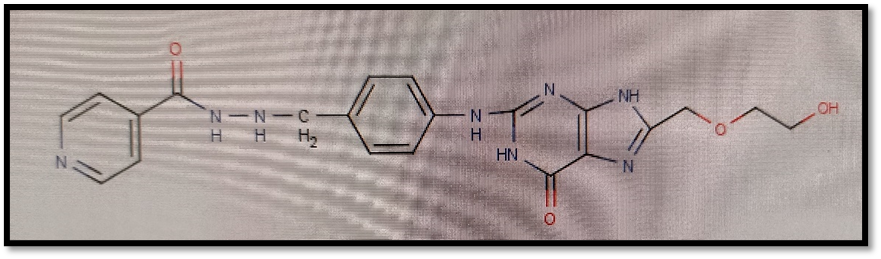
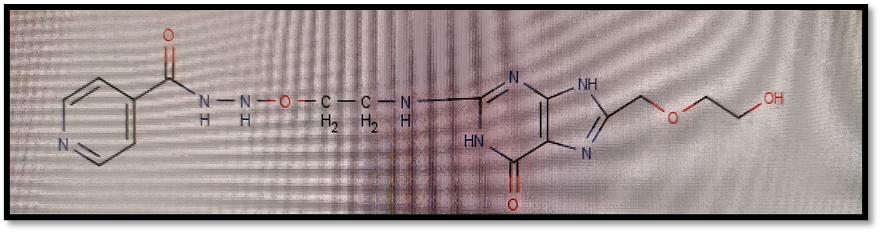
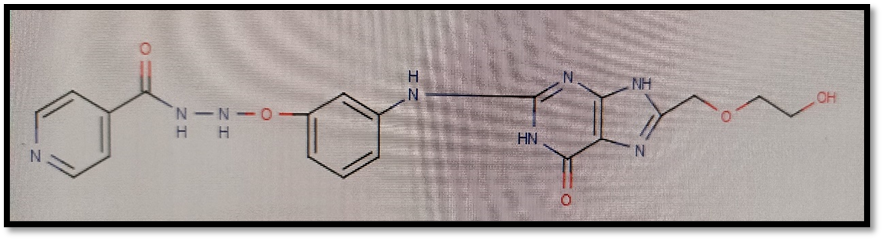
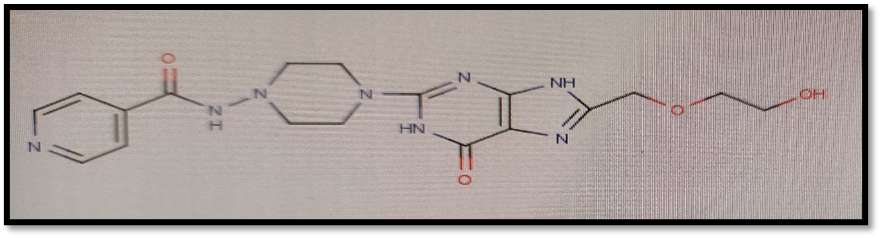
2.1 Selection of Protein For Molecular Docking:
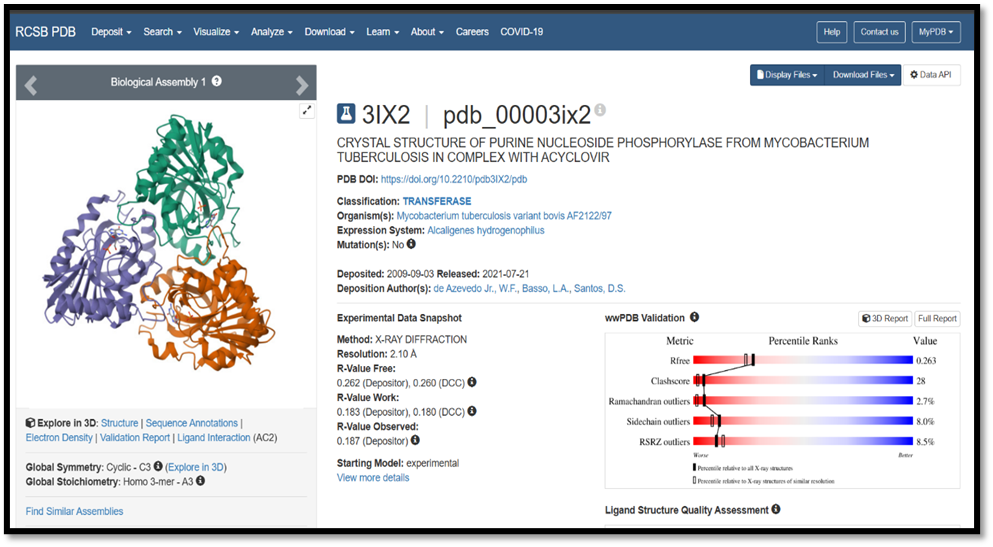
Here isoniazid and acyclovir compounds show possible activity against the adenosine A3 receptor when analysed with Swiss Target Prediction software. We employed the 3IX2 protein from the Protein Data Bank (PDB) for the docking parent compounds. The complete docking procedure makes use of the following software programs: Biovia Discovery Studio, Auto Dock Tools, Marvin View, and Marvin Sketch. Molecular docking is a computational method employed in structure-based drug design to forecast how small molecules (ligands) interact with larger molecules (proteins or receptors). By examining binding interactions, it contributes to the creation of therapeutic compounds. Molecular docking in drug design serves multiple purposes: it aids in identifying possible drug candidates by predicting how a drug molecule (ligand) binds to a protein target (receptor), it enhances our understanding of drug-target interactions, and it eventually supports the development of drugs.
2.3 Chemical Database Search:
The protein data bank (PDB) is a database for the three-dimensional structural data of large biological molecules, such as protein and nucleic acid. The software used for chemical database search is Swiss Target Prediction
2.4 Chemical Structure Drawing:
Chemical structure drawing is graphic representation of the molecular structure showing how the atoms are possibly arranged in the real 3D space. The different software’s are used for drawing of chemical structure i.e., Marvin Sketch.
2.5 Chemical Structure Presentation:
The chemical structure of drug determines its physicochemical properties and further determinates its ADMET properties. The drawn chemical structure should have modified to present in docking. The software used for this is Marvin View.
2.7 Ligand Preparation:
2.8 Protein Preparation:
2.9 Preparation of Grid:
2.10 Executing the commands for Docking:
2.11 Docking Analysis:
1. Open docking log file and find RMSD table.
2. Identify runs with highest ranking and minimum binding energy.
3. Analyze docking results in AutoDock Tools.
4. Visualize ligand-receptor interactions and identify hydrogen bonds.
5. Save docked complex and open in Discovery Studio.
6. Visualize ligand binding site atoms and hydrogen bonds.
2.12. ADMET
Step 1. Go to admetSAR (http://Immd.ecust.edu.cn:8000/)
Step 2. Click on “ Predict “
Step 3. Copy the SMILES from the database and paste it in search bar
Step 4. After clicking on “Predict” button, following result is displayed.
RESULT AND DISCUSSION:
|
Sr. no |
IUPAC Name |
Binding Energy |
No. Of Hydrogen |
Structure |
2D Structure |
3D Structure |
|
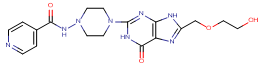
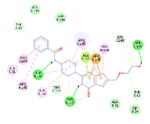
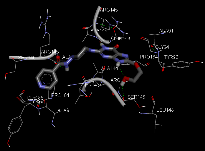
1. |
N-(4-{8-[(2-hydroxyethoxy)methyl]-6-oxo-6,9-dihydro-1H-purin-2-yl}piperazin-1-yl)pyridine-4-carboxymide |
-11.58 |
3 |
|
|
|
|
2. |
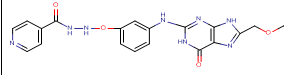
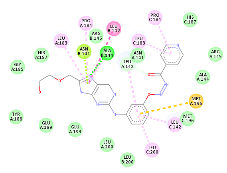
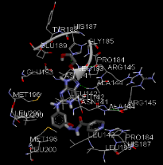
N-[3-({8-[(2-hydroxyethoxy)methyl]-6-oxo-6,9-dihydro-1H-purin-2-yl}amino)phenoxy]pyridine-4-carbohydrazide |
-10.73 |
1 |
|
|
|
|
3. |
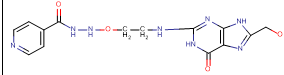
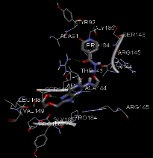
N-[3-({8-[(2-hydroxyethoxy)methyl]-6-oxo-6,9-dihydro-1H-purin-2-yl}amino)ethoxy]pyridine-4-carbohydrazide |
-9.69 |
4 |
|

|
|
|
4. |
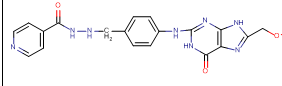
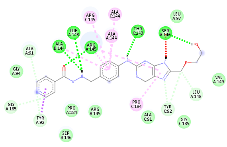
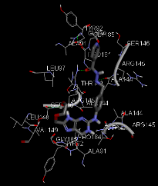
N-[3-({8-[(2-hydroxyethoxy)methyl]-6-oxo-6,9-dihydro-1H-purin-2-yl}amino)phenyl]methyl}pyridine-4-carbohydrazide |
-9.93 |
5 |
|
|
|
According to this table, 1st derivative of isoniazid and acyclovir with binding energy -11.58 occur as most suitable as compare to other 3 derivatives.
CONCLUSION:
According to ADME properties of all 4 derivatives, the derivative 1 with most least binding energy (-11.58) is suitable for docking and synthesis. So, we going to synthesize derivative 1st for our final project.
REFERENCES


Dr. Sonali Banpure*, Bhalekar Pournima, Ingale Pramod, Matade Siddhesh, Mhase Pratiksha, Design, In Silico Evaluation, Synthesis and Biological Evaluation Of 4-Nitroimidazole Derivatives, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 1621-1627 https://doi.org/10.5281/zenodo.15381646
 10.5281/zenodo.15381646
10.5281/zenodo.15381646
