Kasturi Shikshan Sanstha College of Pharmacy, DBATU Shikarpur Pune.
Pharmaceutical drug discovery is an expensive and time-consuming process. The development of a drug from an initial idea to its entry into the market is a very complex process which can take around 5-10 yrs. and cost is very high upto billion. It is an development process involves use of variety of computational techniques, such as structure activity relationship, quantitative structure activity relationship, molecular mechanics, quantam mechanics, molecular dynamics and drug protein docking. The idea for a new development can come from a variety of sources which include the current necessities of the market, new emerging diseases, academic and clinical research, commercial sector. The pharmaceutical industry is under pressure in developing cost effectiveness drug molecule from the previous knowledge and established Quantitative Structure Activity Relationships. The structure-based design is one of reliable and promising techniques used in drug designing. In drug design, the main aim is to find out the three-dimensional structure of pharmacologically significant receptor ligand complexes. The aim of this review is to give an overview on the rational drug design approaches with a case study on drug discovery for influenza A virus, HER2 Receptor, targeting dopamine D3 receptor , purpose , and applications of QSAR. This article highlights the benefits and promises of developing tools for drug discovery.
Drug design is an integrated developing discipline which portends an era of “Tailored drug”.It is an development process involves use of variety of computational techniques ,such as structure activity relationship ,quantitative structure activity relationship(QSAR),molecular mechanics ,quantam mechanics,molecular dynamics and drug protein docking. The QSAR establish a statistical relationship between biological activity or environmental behaviour of the chemicals of interest and their structural properties. QSAR predict chemical behaviour of directly from chemical structure and stimulate adverse effect in cells ,tissues and lab animals minimizing the need to use animals test to comply with regulatory requirements for human health and ecotoxicology. Disposition of drugs in individual region of biosynthesis is one of the main factors determining the place, mode and intensity of their action .The biological activity may be “Positive” as in drug design or “Negative” as in toxicology .Drug design frequently but not necessarily relies on computer modelling is often referred to as “Computer aided drug design”. Drug design that relies on the knowledge of three dimensional structure of the biomolecular target is known as “structure based drug design”. Drug design is the inventive process of finding new medications based on the knowledge of a biological target. despite advances in biotechnology and understanding of biological systems ,drug discovery is still lengthy ,costly ,difficult ,and inefficient process with a high attrition rate of new therapeutic discovery. In the most basic sense ,drug design involves the design of molecules that are complementary in shape and charge to molecular target with which they interact and bind .Drug development and discovery includes preclinical research on cell based and animals models and clinical trials on humans ,and finally move forward to the step of obtaining regulatory approval in order to market the drug.The drug is most commonly an organic small molecules that activates or inhibits the function of a biomolecules such as a protein (receptor and enzymes ),which in turn results in a therapeutic benefit to the patient .In the most basic sense ,drug design involves the design of small molecules that are complementary in shape and change to the biomolecular target with which the interact and therefore will bind to it.

Fig.1
Principles of Drug design:
Lipinski’s Rule of Fives: Lipinski’s rule also known as the Pfizer rule of five or simply the rule of five is a rule of thumb to evaluate drug likeness or determine chemical compound with a certain pharmacological or biological activity has properties that would make it alikely orally active drug in humans .The rule was formulated by Christoper in 1997 ,based on the observation that most medication drugs are relatively small and lipophilic molecules. The rule describes molecular properties for drugs pharmacokinetics in the human body including their ADME .Hence ,the rule does not predict if a compound is a pharmacologically active. The rule is important to keep in mind during drug discovery when a pharmacologically active lead structure is optimized stepwise to increase the activity and selectivity of the compound as well as to ensure drug like physicochemical properties are maintained as describes by Lipinski’s rule . Components of the Rule:
1)Not more than 5 hydrogen bond donors.
2)Not more than 10 hydrogen bond acceptors.
3)A molecular mass less than 500 daltons.
4)An alcohol water partition coefficient (log p)not more than 5.
Note that all numbers are multiplies of five which is the origin of the rules name. The Lipinski’s Rule of Five states that an orally bioavailable drug must neither possess more than five hydrogen bond donors nor more than 10 hydrogen bond acceptors ;its molecular weight must not be grater than 500 g/mol and its log p value not greater than 5 .

Fig.2
QSAR (Quantitative Structure Activity Relationship):
Quantitative structure activity relationship (QSAR)studies represent a non experimental part of drug design encompassing the study of both structure activity and structure property relations in broad sense. The most commonly used mathematical techniques in classical quantitative structure activity relationships (QSAR) work is multiple regression analysis. QSAR is an intellectual exercise of assembling , manipulating , and examining data obtained from physical , chemical, and biological experiments, and correlating them to biological activity . Biological activity of a drug depends on the types and magnitude of interactions between the receptor and the drug molecule . Various structural attributes of a drug molecule, such as electronic distribution , steric features etc. ,are the determining factors regulating the interactions . Parameters must be properties that are capable of being represented by a numerical value. These values square measure wont to manufacture a general equation relating drug activity with the parameters. The goals QSAR studies include a better understanding of the modes of actions, prediction of newer analogs with better activity , classification of active /inactive compounds and optimization of the lead compound to reduce toxicity and increase selectivity. The main properties of a drug that seem to influence its activity measure its lipophilicity, the electronic effects within the molecule and the size and shape of the molecule. Lipophilicity is a measure of a drug’s solubility in lipid membranes. This is usually an important factor in determining how easily a drug passes through lipid membranes. It is used as a live of the convenience of distribution of a drug to its target website. The parameters commonly used to represent these properties are partition coefficients and lipophilic substitution constants for lipophilicity, Hammetts constants for electronic effects and tafts Es steric constants for steric effects . QSAR derived equations take the general form
Biological activity = Function (parameter)
In which the activity is normally expressed as log [1/(concentration term)]where the concentration term is usually C the minimum concentration required to cause a defined biological response .QSAR studies square measures ordinarily applied on teams of connected compounds. However, QSAR studies on structurally diverse sets of compounds are becoming more common. In both instances it is important to consider as wide range of parameters as possible.
The different parameters are:
1)The lipophilic parameters
A. Partition coefficient(P)
B. Lipophilic substituent constants (p)
C. Distribution coefficients
2)Electronic parameters
A. The Hammett constant(s)
3)Steric parameters
4)The Taft steric parameter(ES)
Hansh Analysis :
Hansh and co workers in the early 1960s proposed a multiparameter approach to the problem based on the lipophilicity of the drug and the electronic and steric influences of groups found in its structure. They realised that the biological activity of a compound is a function of its ability to reach and bind to its target site.Hansh planned that drug action may well be divided into 2 stages: 1.The transport of the drug to its site of action. 2.The binding of the drug to the target site. Hansh postulated that the biological activity of a drug may well be associated with all or a number of these factors by straight forward mathematical relationships supported the final format : Log 1/C1/4 k1[partition parameter]+k2[electronic parameter]+k3[steric parameter]+k4
Where, C is the minimum concentration required to cause a specific biological response and k1, k2, k3, k4 are numerical constants obtained by feeding the data into a suitable computer statistical package.
Achievements of QSAR:
1)Forecasting of biological activity.
2)selection of proper substituent’s.
3)Bioisosterism.
4)Drug receptor interaction.
5)Pharmacokinetics information’s.
6)Time saving in synthesis process.
7)NO need to synthesis of derivatives liabrary.
Limitation of QSAR:
1)Regression analysis
2)Improper conditions of biological testing
3)Multiple mode of actions
QSAR methodology: QSAR methodologies have the potential of decreasing substantially the time and effort required for the discovery of new medicines A major step in constructing the QSAR models is to find a set of molecular descriptors that represents variations of the structural properties of the molecule. The QSAR analysis employs statistical methods to derive quantitative mathematical relationship between chemical structure and biological activity. The process of QSAR modelling can be divided into three stages: development, model validation and application.
Application:
1) The application of QSAR models depends on statistical significance and predictive ability of the models.
2) The prediction of a model response using QSAR is valid only if the compound being predicted is within the applicability domain of the model.
3) The applicability domain is a theoretical region of the chemical space, defined by the model descriptors and model response and thus by the nature of the training set molecules.
4) It is possible to check whether a new chemical lies within applicability domain using the leverage approach.
5) A compound will be considered outside the applicability domain when the leverage values is higher than the critical value of 3p/n, where p is the number of model variables plus 1 and n is the number of objects used to develop the model.
CADD (Computer Aided Drug Design):
Computer-aided drug design (CADD) provides a variety of tools and techniques that assist in the various stages of drug design, thereby reducing the cost of drug research and development time. Drug discovery and the development of a new drug is a long, complex, costly and highly risky process that has no equal in the commercial world. Therefore, computer-aided drug design (CADD) approaches are widely used in the pharmaceutical industry to speed up the process. The cost advantage of using computational tools in the lead optimization phase of drug development is significant. The cost and time invested by pharmacological research laboratories are heavy at various stages of drug discovery, starting from therapeutic target setting candidate drug discovery to evaluating the efficacy and safety of newly developed drugs, drug optimization through preclinical and extensive clinical trials. Major pharmaceutical companies have invested heavily in routine ultra High Throughput Screening (uHTS) of large numbers of drug-like molecules. In parallel, drug design and optimization are increasingly using computers for virtual screening. Recent advances in DNA microarray experiments are discovering that thousands of genes involved in a disease can be used to gain in-depth information about disease targets, metabolic pathways, and toxicity of drugs. Theoretical tools include empirical molecular mechanics, quantum mechanics, and more recently statistical mechanics. This latest advance allowed the inclusion of overt solvent effects.
Fig.3
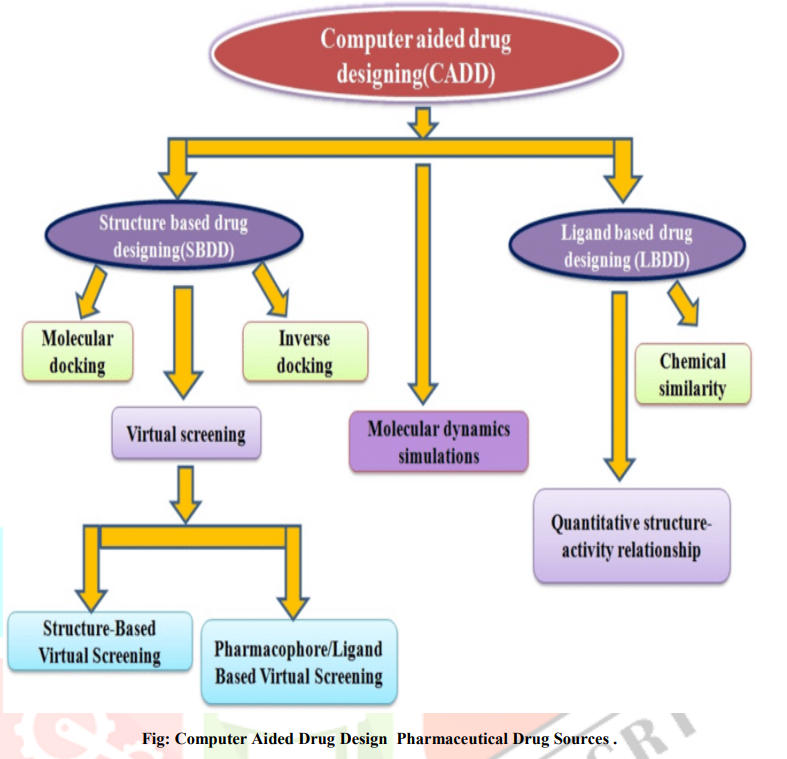
There are two major types of CADD:
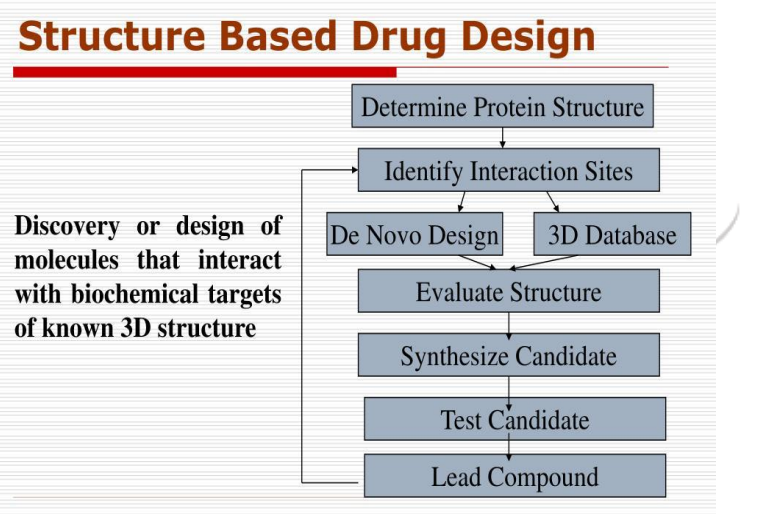
1)Structure Based Drug Design:
The structure of the target protein is known in structure based drug design (SBDD).These methods are very efficient and alternative approach to the discovery and development of drug design course .The (3D)structure of proteins are provided in SBDD. The availability of 3D structures of therapeutically important proteins favours identification of binding cavities and has laid the foundation for structure-based drug design (SBDD). This is becoming a fundamental part of industrial drug discovery projects and of academic researches .
SBDD is a more specific, efficient, and rapid process for lead discovery and optimization because it deals with the 3D structure of a target protein and knowledge about the disease at the molecular level . Among the relevant computational techniques, (SBVS), molecular docking, and molecular dynamics (MD) simulations are the most common methods used in SBDD. These methods have numerous applications in the analysis of binding energetics, ligand–protein interactions, and evaluation of the conformational changes occurring during the docking process.
Fig.4
2)Ligand Based Drug Design:
Ligand based drug design is an indirect approach to facilitate the development of pharmacologically active compounds by studying molecules that interact with the biological target of interest. Ligand based drug design methods are useful in absence of an experimental 3D structure. Due to the lack of an experimental structure, the known ligand molecules that bind to the target are studied to understand the structural and physicochemical properties of the ligands that correlate with the desired pharmacological activity of those ligands. Ligand based method may include natural products or substrate analogues that interact with the target molecule yielding the desired pharmacological effect. In some cases, usually in which data pertaining to the 3-D structure of a target protein are not available, drug design can instead be based on processes using the known ligands of a target protein as the starting point. This approach is known as “ligand-based drug design”.
Pharmacophore:
The original concept of the pharmacophore was developed by Paul Ehrlich during the late 1800s. At that time, the understanding was that certain “chemical groups” or functions in a molecule were responsible for a biological effect, and molecules with similar effect had similar functions in common. The word pharmacophore was coined much later, by Schueler in his 1960 book Chemobiodynamics and Drug Design, and was defined as “a molecular framework that carries (phoros) the essential features responsible for a drug’s (pharmacon) biological activity.” The definition of a pharmacophore was therefore no longer concerned with “chemical groups” but “patterns of abstract features.”
Since 1997, a pharmacophore has been defined by the International Union of Pure and Applied Chemistry as: A pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response. The pharmacophore should be considered as the largest common denominator of the molecular interaction features shared by a set of active molecules. Thus a pharmacophore does not represent a real molecule or a set of chemical groups, but is an abstract concept. Despite this clear definition, the term pharmacophore is often misused by many in medicinal chemistry to describe simple yet essential chemical functionalities in a molecule (such as guanidine or sulfonamides), or common chemical scaffolds (such as flavones or prostaglandins). Often the long definition is simplified to “A pharmacophore is the pattern of features of a molecule that is responsible for a biological effect,” which captures the essential notion that a pharmacophore is built from features rather than defined chemical groups.
Chemical Structure Drawing :
Objectives:
After completing this section, you should be able to,
1)Propose one or more acceptable Kekule structures for any given molecular formula.
2)write the molecular formula of a compound , given its Kekule structure.
3)Draw the short hand structure of a compound , given its Kekule structure.
4)Interpret short hand structures and convert them Kekule structures.
5)Write the molecular formula of a compound , given its short hand structure.
Through general chemistry, you may have already experienced looking at molecular structures using Lewis structures. Because organic chemistry can involve large molecules it would be beneficial if Lewis structures could be abbreviated. The three different ways to draw organic molecules include Kekulé Formulas, Condensed Formulas, and Skeletal structures (also called linebond structures or line formulas). During this course, you will view molecules written in all three forms. It will be more helpful if you become comfortable going from one style of drawing to another, and look at drawings and understanding what they represent. Developing the ability to convert between different types of formulas requires practice, and in most cases the aid of molecular models. Many kinds of model kits are available to students and professional chemists, and the beginning student is encouraged to obtain one. Kekule formulas is just organic chemistry's term for Lewis structures you have previously encountered. In condensed structural formulas, the bonds to each carbon are omitted, but each distinct structural unit (group) is written with subscript numbers designating multiple substituents, including the hydrogens. Line formulas omit the symbols for carbon and hydrogen entirely (unless the hydrogen is bonded to an atom other than carbon). Each straight line segment represents a bond, the ends and intersections of the lines are carbon atoms, and the correct number of hydrogens is calculated from the tetravalency of carbon. Non-bonding valence shell electrons are omitted in these formulas.
1)Kekule (Lewis Structures):
A Kekulé Formula or structural formula displays the atoms of the molecule in the order they are bonded. It also depicts how the atoms are bonded to one another, for example single, double, and triple covalent bond. Covalent bonds are shown using lines. The number of dashes indicate whether the bond is a single, double, or triple covalent bond. All atom labels are shown and all lone pairs are shown.
2)Condensed Formula:
A condensed formula is made up of the elemental symbols. Condensed structural formulas show the order of atoms like a structural formula but are written in a single line to save space and make it more convenient and faster to write out. The order of the atoms suggests the connectivity in the molecule. Condensed structural formulas are also helpful when showing that a group of atoms is connected to a single atom in a compound. When this happens, parenthesis are used around the group of atoms to show they are together. Also, if more than one of the same substituent is attached to a given atom, it is show with a subscript number. An example is CH4, which represents four hydrogens attached to the same carbon. Condensed formulas can be read from either direction and H3C is the same as CH3, although the latter is more common.
Eg.1) CH3CH2OH ,
2)ClCH2CH2CH(OCH3)CH3
3)line formula:
Line angle formulas imply a carbon atom at corners and ends of lines. Each carbon atom is understood to be attached to enough hydrogen atoms to give each carbon atom four bonds. Because organic compounds can be complex at times , line angle formulas are used to write carbon and hydrogen atoms more efficiently by replacing the letter “C” with lines.A carbon atom is present wherever a line intersects another line. Hydrogen atoms are omitted but are assumed to be present to complete each of carbon’s four bonds Hydrogens that are attached to elements other than carbon are shown.Atoms labels for all other elements are shown . lone pair electrons are usually omitted. They are assumed to be present to complete the octet of non carbon atoms. Line formulas help show the structure and order of the atoms in a compound. Combinatorial Chemistry and HTS: Combinatorial chemistry involves the rapid synthesis or the computer simulation of a large number.
Docking:
Docking is a small molecules to protein binding sites was pioneered during early 1980 and remains a highly active area of drug research .When only the structure of a target and its active site is available high throughout docking is primarily used as a hit identification tool.
Molecular Docking:
The computational schemes that attempt to find the best matching between two molecules such as “a receptor and a ligand”.The subject of docking is the formation of non covalent complexex.

Fig. 5 A best match between a protein and a ligand molecule.
Types of Docking:
Blind docking is the docking to homology models of a target ,where the position of active site is assumed to be similar to one in a template protein .Docking to models of transmembranes proteins ,such as G-proteins coupled receptors. Blind Docking refers to docking a ligand to the whole surface of a protein without any prior knowledge of the target pocket. Blind docking involves several trials/runs and several energy calculations before a favourable proteinligand complex pose is found. However, the number of trials and energy evaluations necessary for a blind docking job is unknown. In this type active site of the protein is not known and search for the binding site and subsequently the binding mode ligand is required. It is important for investigating protein protein interactions . Blind Docking refers to docking a ligand to the whole surface of a protein without any prior knowledge of the target pocket. Blind docking involves several trials/runs and several energy calculations before a favorable protein-ligand complex compose is found
Fig.6
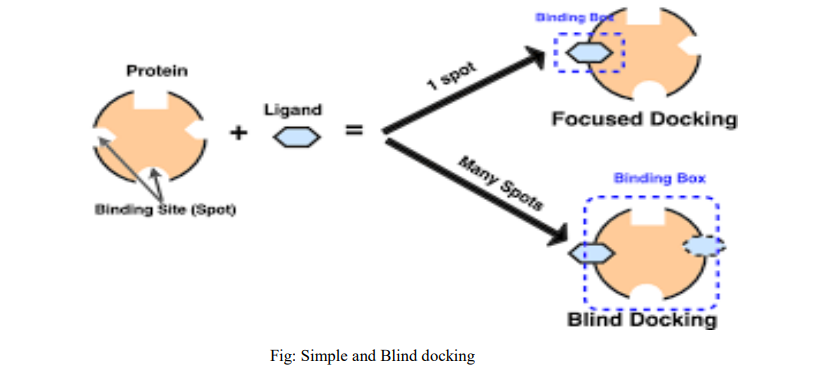
2)Direct docking:
If the active site of the binding is known from X-ray diffraction or from NMR studies ,docking into the known active is called a ‘direct docking’. During direct docking .certain factors such as presence of cofactors ,discrete crystal molecules of water, and catalytic metal ions in the active site of protein or ionized states of the compounds aswell as the effects of the pH ,induced fit and conformational changes of protein must be taken into account if they are participating in protein-ligand interaction.
ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity.):
ADMET Evaluation function module is composed of a series of high-quality prediction models trained by multi-task graph attention framework. It enables the users to conveniently and efficiently implement the calculation and prediction of 17 physicochemical properties, 13 medicinal chemistry measures, 23 ADME endpoints, and 27 toxicity endpoints and 8 toxicophore rules (751 substructures), thereby selecting promising lead compounds for further exploration. Acyclic diene metathesis polymerization (ADMET) is a step growth polycondensation reaction which releases ethylene as a by product. Elimination of ethylene by applying vacuum to the reaction vessel is the driving force of the reaction. Polymerization occurs either in bulk at high temperature up to 190 °C or with the use of solvents at considerably lower temperatures via metathesis using an appropriate catalyst. The first well-defined metathesis catalyst was created by Schrock in 1986 based on tungsten after which molybdenumcontaining Schrock catalysts became prevalent. Both are most effective for producing high molecular weight polymers with degrees of polymerization higher than 150 for hydrocarbon polymers, while being less useful for polymers containing functional groups. Catalysts containing late transition metals, primarily ruthenium, developed by Grubbs and coworkers are best used when functional groups are present; their use has expanded metathesis chemistry considerably. The first-generation ruthenium catalyst, known as Grubbs’ first-generation, led to a variety of new ruthenium structures featuring diverse reactivity and functional group tolerance, while maintaining low rates of olefin isomerization and consequently, chain walking. shows the structures of Schrock's molybdenum and Grubbs’ first-generation catalyst. ADMET monomers normally are ?,?-dienes that produce unsaturated ADMET polymers. The major advantage of ADMET chemistry is the formation of repeat units with symmetrically disposed functionalities (both hydrocarbon and otherwise) along the polymer backbone, a key feature that is a direct consequence of the monomer symmetry. The unsaturated polymer can be further saturated by exhaustive hydrogenation of the polymer chain. shows a symmetric ?,?-diene that yields an unsaturated ADMET polymer with a generic functionality. Functionality is precisely placed along the polymer backbone by controlling symmetrically the length between terminal olefins. Depending upon the nature of the introduced functionality and the design of the ?,?-diene monomer, a diverse library of saturated and unsaturated ADMET polymers can be obtained with the functionality either incorporated in the polymer chain or pendant along the chain. The first significant ADMET polymerization work appeared in 19914 followed by the creation of diverse polymers containing precisely placed in-chain or pendant functional groups. For example, when the R group is an alkyl chain, the resultant polymer can be seen as a model polyolefin; many other polymers containing sulphur, boron, silicon, and amino acids have been synthesized. The following sections review the synthetic methods used for the preparation of those monomers and polymers, along with discussion of secondary structure elucidation and applications of ADMET polymers as functional materials
Combinatorial Chemistry and HTS:
Combinatorial chemistry involves the rapid synthesis or the computer simulation of a large number of different but often structurally related molecules or materials. In a combinatorial synthesis, the number of compounds made increases exponentially with the number of chemical steps. In a binary light-directed synthesis, 2n compounds can be made in n chemical steps. Combinatorial chemistry is especially common in CADD (Computer aided drug design) and can be done online with web based software, such as molinspiration.
History of Combinatorial Chemistry:
Combinatorial chemistry was first conceived about 15 years ago - although it wasn't called that until the early 1990s. Initially, the field focused primarily on the synthesis of peptide and oligonucleotide libraries. H. Mario Geysen, distinguished research scientist at Glaxo Welcome Inc., Research Triangle Park, N.C., helped jump-start the field in 1984 when his group developed a technique for synthesizing peptides on pin-shaped solid supports. At the Coronado conference, Geysen reported on his group's recent development of an encoding strategy in which molecular tags are attached to beads or linker groups used in solid-phase synthesis. After the products have been assayed, the tags are cleaved and determined by mass spectrometry (MS) to identify potential lead compounds. Although combinatorial chemistry has only really been taken up by industry since the 1990s, its roots can be seen as far back as the 1960s when a researcher at Rockefeller University, Bruce Merrifield, started investigating the solid-state synthesis of peptides 19 . In the past decade there has been a lot of research and development in combinatorial chemistry applied to the discovery of new compounds and materials. This work was pioneered by P.G. Schultz et al. in the mid-nineties (Science, 1995, 268: 1738- 1740) in the context of luminescent materials obtained by code position of elements on a silicon substrate. Since then the work has been pioneered by several academic groups as well as industries with large R&D programs.
Principle of Combinatorial Chemistry:
Combinatorial chemistry is a technique by which large numbers of structurally distinct molecules may be synthesized in a time and submitted for pharmacological assay. The key of combinatorial chemistry is that a large range of analogues is synthesized using the same reaction conditions, the same reaction vessels. In this way, the chemist can synthesize many hundreds or thousands of compounds in one time instead of preparing only a few by simple methodology 4 . In the past, chemists have traditionally made one compound at a time. For example compound A would have been reacted with compound B to give product AB, which would have been isolated after reaction work up and purification through crystallization, distillation or chromatography.
Concepts of Combinatorial Chemistry and Combinatorial Technologies:
Combinatorial Technology and Combinatorial Chemistry is a new field joining computer assisted combinatorial chemistry with synthesis of chemical libraries followed by automated screening. The main purpose is to generate thousands structurally diverse compounds as maximizing their diversity, libraries, which are then considered in an experimental screening and synthesis on the basis of their properties.
Combinatorial synthesis on Solid-phase :
Since Merrifield pioneered solid phase synthesis back in 1963, work, which earns him a Nobel Prize, the subject, has changed radically. Merrifield’s Solid Phase synthesis concept, first developed for biopolymer, has spread in every field where organic synthesis is involved. Many laboratories and companies focused on the development of technologies and chemistry suitable to SPS. This resulted in the spectacular outburst of combinatorial chemistry, which profoundly changed the approach for new drugs, new catalyst or new natural discovery.
1) A cross linked, insoluble polymeric material that is inert to the condition of synthesis.
2) Some means of linking the substrate to this solid phase that permits selective cleavage of some or all of the product from the solid support during synthesis for analysis of the extent of reaction(s), and ultimately to give the final product of interest
3) A chemical protection strategy to allow selective protection and deprotection of reactive group
High-throughput screening:
High-throughput screening (HTS) is a method for scientific experimentation especially used in drug discovery and relevant to the fields of biology materials , science and chemistry. Using robotics, data processing/control software, liquid handling devices, and sensitive detectors, high-throughput screening allows a researcher to quickly conduct millions of chemical, genetic, or pharmacological tests. Through this process one can quickly recognize active compounds, antibodies, or genes that modulate a particular biomolecular pathway. The results of these experiments provide starting points for drug design and for understanding the noninteraction or role of a particular location.
Types of High Throughput Screening Assays :
1)Homogeneous assay
2)Heterogeneous assay
3)Biochemical assay
Automation systems:
Automation is an essential element in HTS's usefulness. Typically, an integrated robot system consisting of one or more robots transports assay-microplates from station to station for sample and reagent addition, mixing, incubation, and finally readout or detection. An HTS system can usually prepare, incubate, and analyse many plates simultaneously, further speeding the datacollection process. HTS robots that can test up to 100,000 compounds per day currently exist. Automatic colony pickers pick thousands of microbial colonies for high throughput genetic screening. The term u HTS or ultra-high-throughput screening refers (circa 2008) to screening in excess of 100,000 compounds per day.
Quality control:
High-quality HTS assays are critical in HTS experiments. The development of high-quality HTS assays requires the integration of both experimental and computational approaches for quality control (QC). Three important means of QC are, (i) good plate design, (ii) the selection of effective positive and negative chemical/biological controls, and (iii) the development of effective QC metrics to measure the degree of differentiation so that assays with inferior data quality can be identified. A good plate design helps to identify systematic errors and determine what normalization should be used to remove reduce the impact of systematic errors on both QC and hit selection.
Drugs From Natural Sources:
A natural product is a chemical compound or substance produced by a living organism – found in nature. Natural sources of drug are –plant, animal, mineral, microorganisms. Plant sources are leaf, bark, fruit, seeds. Drugs obtained from animal sources are heparin, insulin, thyroxin, cod liver oil, antitoxic cera. Microorganism sources are bacterial, fungi, molds etc. mineral sources are ferrous sulphate, magnesium sulphate, sodium bi carbonate, aluminium hydroxide. Animal sources are In the broadest sense, natural products include any substance produced by life. the field of organic chemistry, the definition of natural products is usually restricted to mean purified organic compounds isolated from natural sources that are produced by the pathways of primary or secondary metabolism. Many secondary metabolites are cytotoxic and have been selected and optimized through evolution for use as "chemical warfare" agents against prey, predators, and competing organisms. synthetic analogs of natural products with improved potency and safety can be prepared and therefore natural products are often used as starting points for drug discovery. Natural products have served as a major source of drugs for centuries, and about half of the pharmaceuticals in use today are derived from natural products. Interest in natural products research is strong and can be attributed to several factors, including unmet therapeutic needs, the remarkable diversity of both chemical structures and biological activities of naturally occurring secondary metabolites. About 40% of the drugs used are derived from natural sources. Plants provide a fertile source of natural products many of which are clinically important medicinal agents. Different strategies will result in a herbal medicine or in an isolated active compound. Drug discovery scientists often refer to these ideas as "leads," and chemicals that have desirable properties in lab tests are called lead compounds. Pharmaceutical chemists seek ideas for new drugs not only in plants, but in any part of nature where they may find valuable clues. India is the biggest supplier of licit demand for opium required primarily for medicinal purposes. Besides this, India is located close to
CONCLUSION:
Drug design is the creative process of finding new remedies based on the knowledge of biological target This review discusses principle of drug design, various approaches of drug design , lead discovery , lead modification and various types of drug discovery . Bio-isosterism is an important lead modification approach that has been shown to be useful to attenuate toxicity or to modify the activity of a lead , and may have a significant role in the alteration of pharmacokinetics of a lead.
REFERENCES



Chaitali Ingawale*, Sandhya Khomane, Rupali Kharat*, Shrushti Uchale, Computer Aided and AI based Drug Design, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 2222-2234. https://doi.org/10.5281/zenodo.14498665
 10.5281/zenodo.14498665
10.5281/zenodo.14498665
