Ideal Institute of Pharmacy Posheri, Wada, Tel -Wada, Dist. - Palghar, State - Maharashtra.
BACKGRAOUND: he integration of artificial intelligence (AI) into natural product research represents a rapidly evolving domain with the capacity to transform drug discovery. Recognizing AI's promising role in advancing the investigation and development of natural compounds, this study seeks to examine how AI technologies are being applied to enhance research and innovation in this field. Conventional trial-and-error methods in herbal drug research are increasingly being replaced by data-driven strategies through the integration of AR Artificial intelligence has revolutionized phytochemical research by streamlining the analysis of complex 'omics' datasets and accelerating the identification of new metabolites, structural characterization, and comprehensive metabolite profiling in plantstificial intelligence. METHODE : "This study conducted an extensive analysis of how artificial intelligence is utilized in the research and development of natural products." RESULT : AI has significantly transformed the process of discovering and developing natural products. By streamlining the analysis of vast datasets, AI systems can pinpoint promising new compounds with greater speed and precision compared to conventional techniques.
Natural compounds sourced from plants, microorganisms, and marine life have played a foundational role in drug development throughout history. These substances have been instrumental in creating vital medications such as antibiotics, cancer therapies, and drugs that suppress immune responses. Their wide-ranging chemical structures and distinctive biological effects make them essential tools in tackling diverse health issues (1,2) Penicillin, sourced from the Penicillium mold, transformed how bacterial infections are treated, while paclitaxel, obtained from the Pacific yew tree, represented a significant advancement in cancer treatment. (3)
Phytochemicals, which are naturally occurring compounds in plants, exhibit a wide range of therapeutic effects. Their use in traditional medicine over centuries underscores their value and suggests strong potential for development into innovative pharmaceutical agents (4) Modern scientific methods, particularly structural and computational biology, provide remarkable new avenues for exploring natural products. Through structural biology, researchers have uncovered the three-dimensional configurations of phytochemicals, which are instrumental in advancing studies using molecular docking and virtual screening to identify novel compounds with pharmacological potential. (5) Cancer remains one of the leading causes of death worldwide, with around 19.3 million new cases and nearly 10 million fatalities reported in 2020. Although advancements have been made in treatments such as chemotherapy, radiotherapy, and immunotherapy, their overall effectiveness is still hindered. This is largely due to challenges like multidrug resistance (MDR), widespread toxicity, and poor bioavailability of many anticancer medications. (6)
2.TRADITIONAL PHYTOCHEMICAL SCREENING APPROCHES:
"Artificially created compounds have played a leading role in medicinal chemistry. (7) "Nevertheless, due to their diverse bioactivities, phytochemicals are increasingly considered promising alternatives for new drug development (8)
Phytochemicals such as alkaloids, terpenes, and flavonoids serve as promising sources for lead compound development. (9) Their significance in drug discovery is underscored by their vast chemical diversity, wide-ranging biological activities, and longstanding use in traditional medicine. (10) Consequently, screening phytochemicals plays a crucial role in identifying potential drug candidates.
Modern drug discovery employs advanced techniques like high-throughput screening, structure-based drug design, and computational modeling(11) Phytochemicals can be chemically modified to improve therapeutic properties, making them valuable assets for creating personalized treatment strategies. While synthetic compounds have traditionally dominated drug development, natural products are increasingly being investigated. However, discovering new drugs from natural sources remains challenging due to their intricate structures and the complexities involved in their extraction and identification. (12)
2.1 Traditional vs. Modern Approaches in Drug Discovery
Pharmacological research has long been committed to uncovering new compounds that can effectively treat a wide range of diseases. Historically, this pursuit has involved methods such as rational drug design—where synthetic molecules are crafted based on existing drugs—and ethnopharmacology, which draws on traditional remedies used by indigenous cultures. Another widely used approach involves harnessing natural substances derived from plants and animals (13). While these strategies have yielded beneficial outcomes in some cases, they often require extensive time and effort to produce results.
However, with the rise of structural and computational biology, the exploration of phytochemicals for drug development has reached unprecedented potential. These advanced technologies offer a level of precision and insight into biological mechanisms that surpasses conventional lab-based experiments. Such innovations are proving invaluable in the quest to develop new therapies for the many health challenges facing humanity. (14) Nonetheless, the process of identifying, designing, and validating effective drug candidates remains a complex and demanding endeavor.
2.2Traditional Drug Discovery and the Role of Serendipity
Unplanned discoveries have significantly influenced the development of life-saving drugs. One of the most iconic examples is Alexander Fleming’s accidental identification of penicillin in 1928, when a bacterial culture became unintentionally contaminated. (15)
Another notable case is the development of ivermectin, an antiparasitic medication. This breakthrough emerged from a fortuitous collaboration between Satoshi ?mura, who isolated the bacterium Streptomyces avermitilis from Japanese soil, and William Campbell, who recognized its potential to combat parasitic infections. Initially intended for veterinary use, ivermectin was later approved to treat human diseases such as onchocerciasis and lymphatic filariasis. This discovery highlights the importance of interdisciplinary cooperation and the unexpected nature of scientific progress. ?mura and Campbell were honored with the Nobel Prize in Physiology or Medicine in 2015 for their contributions. (16)
In another instance of serendipity, researchers in 1957 reported the antidepressant effects of iproniazid, a monoamine oxidase inhibitor, at an American Psychiatric Association meeting in Syracuse, New York (17). Originally synthesized in 1951 by Herbert Fox at Roche Laboratories for tuberculosis treatment, iproniazid unexpectedly induced euphoric behavior in some patients, as observed by Orcnstein, Robitzek, and Sclikoff in 1952. This surprising effect, later validated by Zeller, led to its recognition as one of the earliest antidepressants. (18)
2.3 Contemporary Approaches to Drug Discovery
The convergence of molecular biology, biochemistry, and structural biology has revolutionized the field of drug development. (19) One of the most advanced strategies is rational drug design, which relies on a deep understanding of disease mechanisms and the structural and functional characteristics of target molecules. This detailed insight allows scientists to create highly specific and effective therapeutic agents aimed at precise molecular interactions. However, this process demands thorough investigation into both the disease and the target molecule, making it one of the most groundbreaking techniques in modern medicine. (20)
High-throughput screening (HTS) has become a cornerstone in pharmaceutical research, enabling the rapid assessment of vast compound libraries for biological activity against defined targets or disease models. These libraries may include synthetic chemicals, natural extracts, genome-wide gene knockouts, or RNA interference tools. Despite its speed and efficiency, HTS faces challenges such as limited availability of suitable assay materials and potential inaccuracies in results. (21,22)
To complement these methods, computational techniques like molecular modeling and docking are employed to predict how candidate molecules will interact with their targets. These tools also help evaluate the chemical properties of compounds, allowing researchers to prioritize them for further testing ( 23,24) The effectiveness of computational approaches depends largely on the quality and relevance of the input data, as well as the accuracy and robustness of the algorithms used. (25,26)
|
Phytochemical identification Thin-layer chromatography, High-Performance Liquid Chromatography (HPLC), Gas Chromatography (GC), Mass Spectrometry (MS), Nuclear Magnetic Resonance (NMR) Spectroscopy, Ultra-violet visible (UV-Vis) Spectroscopy, Infrared (IR) spectroscopy, bioinformatics |
|
Target prediction/identification (Genomics, transcriptomics, proteomics, chemical genetics, phenotypic screening, bioinformatics and computational methods, structural biology, CRISPR, known phytochemicals from specific plants) |
|
Target confirmation (Gene manipulation, RNA interference-RNAi, analysis, use of modulating compounds, patient derived cells and organoids, High Throughput Screening (HTS), pharmacological validation, in silico validation, protein interaction analysis-reference proteins can be obtained from the PDB) |
|
Lead identification (High-throughput screening (HTS), fragment-based lead discovery (FBLD), structure-based drug design (SBDD), phenotypic screening, virtual screening, natural product screening biomimetic synthesis, combinational chemistry |
|
Lead optimisation (Structure-based design (SBDD), quantitative structure-activity relationship (QSAR) modelling, in vitro testing, ADME testing, toxicity testing, physicochemical and pharmacodynamics (PK/PD) modelling, parallel synthesis and combination chemistry) |
|
Lead optimisation (Structure-based design (SBDD), quantitative structure-activity relationship (QSAR) modelling, in vitro testing, ADME testing, toxicity testing, physicochemical and pharmacodynamics (PK/PD) modelling, parallel synthesis and combination chemistry) |
|
Preclinical testing (in vitro studies and in vivo studies on appropriate disease models, pharmacokinetic studies, pharmacodynamic studies, safety pharmacology, genotoxicity, and carcinogenicity studies, reproductive toxicology, immunotoxicology |
|
Lead optimisation. (Structure-based design (SBDD), quantitative structure-activity relationship (QSAR) modelling, in vitro testing, ADME testing, toxicity testing, physicochemical and pharmacodynamics (PK/PD) modelling, parallel synthesis and combination chemistry) |
|
Preclinical testing (In vitro studies and in vivo studies on appropriate disease models, pharmacokinetic studies, pharmacodynamic studies, safety pharmacology, genotoxicity, and carcinogenicity studies, reproductive toxicology, immunotoxicology |
|
Clinical trials Phase I, II, III and IV trials |
|
Approval (Submission of Marketing Authorisation Application, review by regulatory agencies, inspections, advisory committee review, decision |
2.4 Fig 1: 1 sequential stages phytochemical drug discovery and development
3.Ovarvieww phytochemical screening approaches
Computational phytochemistry integrates advanced mathematical and computational tools to enhance phytochemical research, from biosynthetic pathways to molecular modeling. Techniques like Density Functional Theory (DFT) play a pivotal role in predicting molecular properties and electronic structures. (27)
Here's a deeper look into the scope and significance of computational phytochemistry
3.1 What Is Computational Phytochemistry?
Computational phytochemistry refers to the use of computational, mathematical, and statistical methods to analyze, predict, and simulate phytochemical phenomena. It bridges theoretical models with experimental data, enabling researchers to explore plant-derived compounds more efficiently and accurately.
3.2 Key Applications
Density Functional Theory (DFT) has long been a cornerstone in computational chemistry, especially in phytochemical studies where understanding molecular behavior is crucial. Before the rise of AI, DFT provided a robust framework for:
In their 2014 study, Ullah and colleagues conducted a comparative theoretical and experimental analysis of Pistagremic acid, a bioactive compound. Key aspects of their work include:
3.5 Why This Matters
This study exemplifies how DFT bridges theoretical chemistry and experimental validation, offering insights into molecular behavior that are essential for drug discovery, natural product analysis, and bioactivity profiling.
Would you like help paraphrasing or expanding this into a formal academic paragraph or integrating it into a larger literature review?
Dynamic techniques can be employed to elucidate the molecular structure of phytochemicals dissolved in organic solvents, offering complementary insights to those obtained from NMR spectroscopy. In a theoretical study, the ^1H NMR spectrum of 5,4′-Dihydroxy-7,5′,3′-trimethoxyisoflavone—isolated from Ouratea ferruginea tea—was analyzed using the hybrid B3LYP density functional theory (DFT) method (Hernandes et al., 2020). (30)
4. Computer-Aided Structure Elucidation (CASE) in Phytochemistry
Computer-assisted methods for predicting and determining molecular structures have been in use for over five decades, originally developed using spectroscopic data. Today, CASE primarily relies on one-dimensional (1D) and two-dimensional (2D) nuclear magnetic resonance (NMR) data, which significantly reduce errors in identifying the structures of phytochemicals.
CASE operates on a set of axioms that link molecular fragments to specific spectral features. For example, if a molecule contains a certain fragment (e.g., AI), its presence can be inferred from characteristic spectral regions. However, when using 2D-NMR data, a hypothesis based on general spectral patterns tends to be more effective than rigid axioms.
The structural backbone of a phytochemical is typically derived from key 2D-NMR techniques such as COSY and HMBC. Final structure elucidation involves assembling:
These fragments are validated against known axioms to build a complete molecular structure.
4.1. CASE Software Tools
These tools enhance the accuracy and efficiency of phytochemical structure elucidation (31)
Fig. 2 Outline of computer-assisted structure elucidation (CASE) of phytochemicals. The data of phytochemical col lected from the spectroscopic instruments will be preprocess
Group assignments were determined using HMBC correlations, followed by a comparison with predicted ^13C NMR data generated through the ACD/Spectrus Processor (Du et al., 2019) (32). Another significant computational approach is chemometrics, which extracts essential chemical insights from experimental data using mathematical, statistical, and algorithmic techniques. The chemometric workflow typically includes experimental design, data preprocessing, classification, calibration, knowledge extraction, and interpretation (Sarker and Nahar, 2024) (33). Notably, chemometrics employs principal component analysis (PCA) and hierarchical cluster analysis (HCA) to provide both descriptive and predictive insights. PCA is an unsupervised method used for recognizing patterns in multivariate datasets, whereas HCA groups data based on similarity measures to derive meaningful outcomes (Patras et al., 2011). (34)
Machine learning-enhanced analytical methods are transforming phytochemical (35) research,
offering powerful avenues to investigate the concealed molecular characteristics of plant-derived samples. These advanced techniques serve as gateways to decode complex phytochemical profiles, enabling researchers to identify, classify, and predict the bioactivity of natural compounds with greater precision and efficiency.
4.2 Table 1 : Representative Tools in Current Phytochemical Research (35)
While the exact contents of Table 1 may vary by publication, based on recent literature, it typically includes the following categories of tools and techniques:
|
Tool/Technique |
Purpose |
|
Chromatography (HPLC, GC) |
Separation and quantification of phytochemicals |
|
Mass Spectrometry (MS) |
Molecular weight determination and structural analysis |
|
Nuclear Magnetic Resonance (NMR) |
Detailed structural elucidation of organic compounds |
|
Spectroscopy (UV-Vis, IR) |
Detection of functional groups and compound identification |
|
Metabolomics Platforms |
Comprehensive profiling of metabolites in plant samples |
|
AI/ML Algorithms |
Pattern recognition, predictive modeling, and data integration |
|
HerbIntel Platform |
AI-powered tool for decoding plant-based molecular structures |
|
Omics Data Integration Tools |
Combine genomics, proteomics, and metabolomics for holistic analysis |
4.3 Table 2: Plant Databases and Their Applications (36)
|
Data base |
Source |
Application |
|
Ensembl plant |
Ensemble plant |
Genome annotation |
|
NCBL taxonomy |
NCBL Taxonomy |
Taxonomy |
5. Application of Al phytochemical screening :
1. Artificial intelligence has revolutionized the discovery of herbal medicines by optimizing key stages such as target identification, deorphanization, metabolome exploration, synthesis design, and virtual screening. The integration of deep learning (DL) facilitates the analysis of biosynthetic gene clusters and metabolic pathways. Additionally, techniques like pharmacophore modeling, molecular docking, and bioactivity fingerprinting enhance the accuracy of identifying molecular targets. (37,38)
2. Through algorithmic prediction of toxicity, bioactivity, and pharmacokinetics, artificial intelligence reveals hidden patterns within large datasets to identify promising bioactive compounds. Moreover, AI—especially machine learning (ML) and deep learning (DL) techniques—facilitates automatic compound recognition and comparison with known molecules during dereplication. This is achieved by leveraging spectroscopic data and convolutional neural networks (CNNs). (39)
3. AI accelerates de novo drug development by leveraging in silico screening techniques and reinforcement learning to optimize multiple aspects of drug efficacy. It also enhances the reliability of natural therapies through robust quality control frameworks built on machine learning pipelines. These include image-based authentication using convolutional neural networks (CNNs) such as ResNet101 and Inception V3, along with real-time detection of adulterants. (40)
4. Meanwhile, the analysis of synergistic effects in polyherbal formulations—such as Diabet and Dihar, which act as hypoglycaemic agents—is greatly facilitated by deep learning models. Tools like DeepSynergy, SynergyFinder, and network pharmacology approaches enable the prediction of safer and more effective herbal combinations." (41)
5. By enhancing the pharmacokinetic properties of promising bioactive compounds such as quercetin, kaempferol, and vancomycin, the integration of artificial intelligence with natural product research is advancing precision medicine—enabling the development of personalized, cost-effective therapies for a wide range of diseases (42)
6. Advancements in computational omics technologies have significantly expanded opportunities for drug discovery, particularly by uncovering diverse natural product sources. At the same time, progress in computational drug design—especially through artificial intelligence methods like machine learning—has enhanced the ability to predict biological activity and design novel compounds aimed at specific molecular target (43)
7. Artificial intelligence has significantly impacted computer-aided drug discovery, with its influence growing due to the widespread adoption of machine learning—particularly deep learning—across various scientific fields. This progress is further propelled by continual improvements in computing technologies, both hardware and software. Despite initial apprehensions that AI might supplant traditional pharmaceutical innovation, medicinal chemistry has ultimately gained from its integration into the drug discovery process. (44)
8. Natural compounds derived from fungi, bacteria, plants, animals, and other organisms serve as an important foundation for modern drug discovery. Their rich structural variety and biological significance make them excellent candidates for initiating new drug development. In this context, computational methods play a crucial role, either as a preliminary step or as a complement to laboratory-based testing (45)
9. Machine learning has significantly transformed the pharmaceutical industry, with both supervised and unsupervised techniques being applied across various phases of drug development. Clustering methods have been instrumental in tasks such as segmenting cell-type images, predicting the druggability of protein targets, and designing new molecules from scratch. Supervised learning models, including regression and classification algorithms, have helped identify promising therapeutic targets for conditions like Huntington's disease. Additionally, these models have been used to predict biological activity and assess key pharmacokinetic and toxicological properties—namely absorption, distribution, metabolism, excretion, and toxicity (ADME/Tox) (Vamathevan et al. (46)
10. Natural product research, a reliable foundation for modern small molecule drug discovery, has increasingly embraced computational approaches powered by artificial intelligence and machine learning. In the early 2000s, techniques like principal component analysis and self-organizing maps were primarily employed to chart the chemical landscape of natural products and convert organic molecules into digital formats. Over the following decade, machine learning-based binary classifiers emerged to predict the biological functions of these compounds. More recently, scientists have started leveraging neural network architectures to aid in molecular design and genome mining. (47)
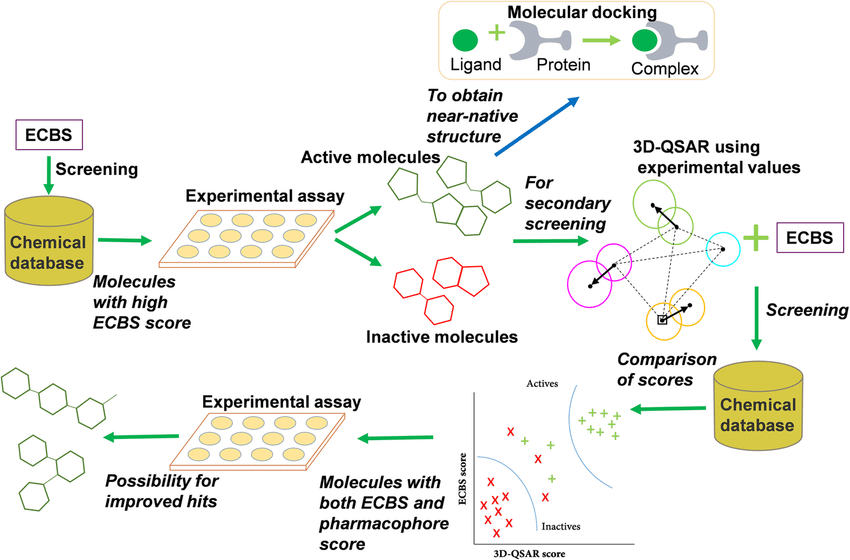
5.1 Fig:3 Compound prediction and virtual screening QSAR models:
5.2 AI has revolutionized QSAR modeling, transforming it from a classical predictive tool into a dynamic, deep learning-powered engine for drug discovery.
Here's a paraphrased version of your sentence with added clarity and flow:
With the advent of artificial intelligence, quantitative structure–activity relationship (QSAR) modeling—an established technique in computer-aided drug design for over six decades—has undergone remarkable advancements, evolving into a more powerful and versatile approach (Tropsha et al., 2024).
To expand on this idea:
5.3 Quantitative Structure–Activity Relationship (QSAR) modelling
is a foundational technique in cheminformatics that explores how molecular characteristics influence chemical, biological, and toxicological behaviors. Traditionally applied in lead optimization during drug discovery, QSAR has evolved to support broader applications, including virtual screening for hit and lead identification, prediction of drug-like properties, and chemical risk assessment. These advancements have been driven by the development of more robust models, improved validation protocols, and a greater emphasis on external validation (Golbraikh et al., 20. (50)
Quantitative Structure–Activity Relationship (QSAR) modeling entails the collection and refinement of large datasets of natural compounds, the computation of molecular descriptors, and the use of machine learning techniques to forecast biological effects and potential toxicities. With the integration of advanced AI methods like deep learning, contemporary QSAR models have demonstrated significant success in pinpointing potential drug candidates with enhanced pharmacological characteristics. This approach has been validated through various successful case studies in the development of improved therapeutics derived from natural products (Kar & Roy, 2012). (51)
In QSAR modeling, the initial phase involves preparing the data. This begins with identifying a pertinent chemical, biological, or toxicological target. Following this, a comprehensive dataset must be assembled and refined, with relevant molecular descriptors selected and computed. The next step is to choose a suitable machine learning algorithm for model development. To ensure the reliability of the analysis, the dataset is repeatedly divided into external validation and modeling subsets. During the model-building stage, the modeling subset undergoes multiple rounds of splitting into training and testing sets to enhance robustness (Wang et al., 2024). (52)
In combinatorial QSAR (combi-QSAR), models are developed using training datasets and assessed with test datasets. This modeling process is repeated for each combination of descriptor sets and computational methods. Models that demonstrate strong statistical reliability are chosen for further external validation. To avoid overfitting and random correlations, a Y-randomization test is performed. The validation phase includes generating consensus predictions for an external test set, ensuring the predictions fall within the model's applicability domain (AD). To refine the process, optimal Z-score thresholds are established based on the accuracy and coverage of the consensus predictions. Subsequently, virtual screening of chemical libraries is carried out by conducting similarity searches using the training or modeling datasets filtered by the Z-threshold. Compounds that pass this filter are then evaluated using the consensus QSAR models (Golbraikh et al., 2017). (53)
6. ROLE OF ARTIFICIAL INTELLIGENCE IN NATURAL PRODUCT RESEARC
6.1 Overview of AI Technologies
Artificial Intelligence (AI) refers to a collection of computational methods designed to process intricate datasets, uncover patterns, and support decision-making. Within the realm of natural product research, AI is revolutionizing the field by streamlining labor-intensive processes and unlocking new possibilities for identifying and developing biologically active compounds. Prominent AI technologies include:
6.2 Virtual Screening and Predictive Modeling
Artificial intelligence has revolutionized drug discovery by enabling virtual screening of extensive chemical databases to identify compounds with promising therapeutic effects. Machine learning (ML) and deep learning (DL) models are employed to predict key attributes such as binding affinity, solubility, and toxicity. These advanced computational tools minimize dependence on conventional trial-and-error approaches, thereby accelerating the drug development process and optimizing resource utilization. (55)
6.3 Structure Elucidation and Activity Prediction
Determining the intricate structures of natural compounds typically involves sophisticated spectroscopic methods. Artificial intelligence plays a pivotal role in this process by interpreting spectral information and forecasting molecular configurations. Among AI approaches, deep learning stands out for its ability to link molecular characteristics with biological functions, thereby supporting the refinement of lead compounds to improve their effectiveness and safety. (56)
6.4 Data Mining and Integration
Research in natural products produces extensive datasets encompassing chemical, biological, and genomic information. Artificial intelligence tools play a crucial role in extracting valuable insights from these datasets to pinpoint potential bioactive compounds. Natural
language processing (NLP) algorithms examine scientific literature, patent documents, and database records to reveal patterns, connections, and innovative prospects. By merging these varied data sources, the process of discovering new drug candidates becomes significantly faster and more efficient. (57)
6.5 AI in Natural Product Research: Transformative Applications
Artificial Intelligence has significantly transformed natural product research by tackling major obstacles and boosting productivity across multiple domains.
Would you like me to expand on the specific areas where AI is making an impact—like drug discovery, compound classification, or biosynthetic pathway prediction? (58)
6.6 Benefits of AI in Natural Product Research
Integrating artificial intelligence into natural product research brings a wide range of advantages:
7. Computational Strategies for Discovering Phytochemical Drug Candidates
Modern computational techniques have proven to be valuable tools in the discovery and refinement of phytochemical-based therapies. Methods such as machine learning, virtual screening, molecular dynamics simulations, and molecular docking have been successfully applied to assess and enhance the biological properties of phytochemicals.(60) Virtual screening is a widely used computational method in drug development that enables the swift assessment and ranking of chemical compounds for potential experimental validation against a particular biological target or disease model.(61) Several approaches can be used, e.g., molecular descriptors and fingerprint-based similarity searching to ligand-based pharmacophore models or structure-based techniques. (62) Several approaches can be used, e.g., molecular descriptors and fingerprint-based similarity searching to ligand-based pharmacophore models or structure-based techniques. (63) Virtual screening methods can be applied to large databases containing known phytochemicals or in-silico-generated libraries mimicking natural products This efficient technique manages large datasets and can reduce the number of compounds evaluated in biological assays (64)
7.1. Molecular docking has revolutionized phytochemical drug discovery by providing a computational approach to forecast how a phytochemical interacts with its target
protein(s)(65) This tool plays a crucial role in identifying phytochemicals with strong potential for further experimental validation. A wide array of computational tools and algorithms have been developed to support this process. Some of the most commonly used platforms include AutoDock Vina, AutoDock GOLD, Discovery Studio, FRED, Glide, ICM, Surflex, MCDock, MOE-Dock, FlexX, DOCK, LeDock, rDock, Cdcker, LigandFit, and UCSF Dock. (66) "Molecular docking plays a crucial role in pinpointing the molecular targets of nutraceuticals, aiding in the treatment of various diseases."(67)
During the COVID-19 pandemic, molecular docking played a pivotal role in identifying phytochemicals with potential to inhibit SARS-CoV-2 replication and pathogenesis.
Molecular docking emerged as a powerful computational tool to screen and evaluate bioactive compounds from medicinal plants against key SARS-CoV-2 proteins, especially the main protease (Mpro), which is essential for viral replication. Here's how it contributed (68)
7.2 Role of Molecular Docking in COVID-19 Phytochemical Research (69)
7.3 Examples of Promising Phytochemicals (70)
7.4 Significance and Impact (71)
This approach not only advanced the search for COVID-19 therapeutics but also reinforced the value of integrating computational biology with ethnopharmacology.
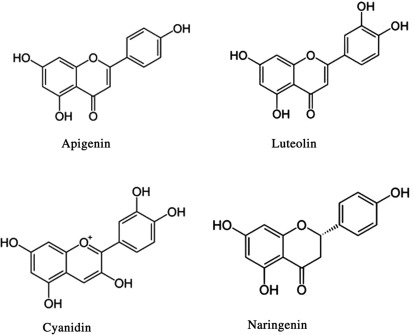
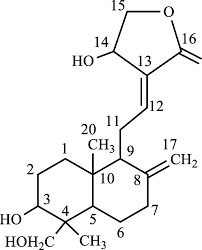
Fig : 4 : Apigenin (72), Luteolin (73), Cyanidin (74), Naeingenin(75) , Andrograpgolide(76)
Andrographolide
"Additionally, other phytochemicals, such as phenolics and terpenoids, have shown potential as leads, including quercetin (23), luteolin (24), and neoandrographolide (25) (Figure 10), that were identified as potential inhibitors of SARS-CoV-2 druggable protein targets. It was shown that their interaction could disrupt viral replication and pathogenesis [13]."(77)
7.5 Molecular Docking in Phytochemical-Based Anticancer Drug Discovery
Molecular docking has played a pivotal role in identifying potential anticancer agents derived from phytochemicals. For example, a study by Swargiary and Mani (2021) identified bayogenin, asiatic acid, and andrographolide as promising lead compounds for targeting Hexokinase 2 (HK2), a key enzyme involved in cancer metabolism. Among these, bayogenin and andrographolide demonstrated the strongest binding affinities to HK2, while asiatic acid also showed interaction, albeit to a lesser extent. These findings suggest that these compounds could serve as novel anticancer agents targeting HK2, pending further validation through in vitro and in vivo studies. (78)
In another investigation focusing on Sauropus androgynus, researchers employed a combination of molecular docking and network pharmacology to uncover key target genes and underlying mechanisms. The study identified AKT1, mTOR, AR, PPID, FKBP5, and NR3C1 as primary targets. (79) Notably, the PI3K-Akt signaling pathway—a crucial regulatory axis in various pathological conditions—was significantly influenced. This integrative approach, combining network pharmacology, molecular docking, and in vitro experiments, provided deeper insights into the anticancer and anti-inflammatory molecular activities of S. androgynus.(80)
8. Case study :
8.1 AI-Driven Approaches and Case Studies Virtual screening :
Virtual screening (VS) is a key computational technique in drug discovery that facilitates the automated analysis of large molecular libraries to pinpoint promising therapeutic candidates. Serving as an initial filtering step, VS efficiently discards compounds with unfavorable characteristics, thereby refining the selection to molecules with a greater potential for biological efficacy (Oliveira et al., 2023) (81)
During the virtual screening process, candidate ligands may be modified in terms of their chemical composition and structural features to enhance their pharmacokinetic properties—specifically absorption, distribution, metabolism, excretion, and toxicity (ADMET). A typical virtual screening pipeline involves two key computational stages. The initial phase focuses on library preparation, which encompasses the collection of compound structures and the conversion of these data into standardized formats suitable for computational analysis, such as SDF, SMILES, and MOL2 ( 82)
The process begins with generating molecular conformers and correcting any stereochemical or valence-related inaccuracies. To identify promising chemical candidates, computational tools are employed in the next phase. Final validation is carried out through experimental methods, including in vitro and in vivo assays such as enzyme inhibition or cell line testing. Over time, artificial intelligence-driven virtual screening has incorporated a wide range of computational strategies. Integrating these digital approaches with laboratory techniques significantly enhances the chances of discovering novel bioactive compounds (Santana et al., 2021(83)
Ligand -based virtual screening (LBVS) and structure-based virtual screening (SBVS) are two computational approaches commonly employed in compound screening. To identify new bioactive molecules targeting specific biological systems or molecular targets, these methods are often integrated into comprehensive virtual screening strategies. LBVS, in particular, predicts molecular activity by analyzing a collection of known bioactive compounds. This technique evaluates various intrinsic properties of the compounds, such as their electronic characteristics, topological features, physicochemical parameters, and structural attributes (Berenger et al., 2017; Garcia-Hernandez et al., 2019). (85)
Computational tools encompass a range of techniques such as machine learning, cheminformatics filters, pharmacophore modeling, similarity-based searches, and quantitative structure–activity relationship (QSAR) analysis. In contrast, structure-based virtual screening (SBVS) utilizes the three-dimensional configuration of a bioreceptor to explore how ligands interact with its binding site. The success of this method hinges on a thorough understanding of intermolecular forces, the makeup of binding site residues, ligand affinity, and the conformational dynamics of the bioreceptor (Maia et al., 2020(87)
Structure-based virtual screening (SBVS) employs several key strategies to enhance ligand binding to bioreceptors. These include molecular docking to predict optimal binding orientations, molecular dynamics simulations to assess the stability and flexibility of ligand-receptor interactions, and structure-based pharmacophore modeling to identify essential features for binding affinity (Wang et al., 2020) (88)
Visual systems (VS) are fundamentally dependent on a robust foundation of knowledge, drawing heavily on both the depth and breadth of available information about the subject being examined. Their effectiveness hinges on the meticulous curation and refinement of this data to ensure accurate and meaningful analysis (Kirchweger & Rollinger, 2018) (89)
Limited availability of 3D libraries for natural products (NPs) has posed a challenge to conducting extensive virtual screening for bioactive compounds. Despite this, several impactful studies have emerged. Liu and Zhou explored marine and traditional Chinese medicine metabolites to identify potential inhibitors of the SARS-CoV protease. Toney et al. discovered sabadinine, a terpenoid alkaloid, as a promising anti-SARS candidate. Moro highlighted ellagic acid as a strong inhibitor of protein kinase CK2. Furthermore, Zhao and Brinton demonstrated the utility of receptor-based molecular docking in pinpointing flavonoid compounds with high affinity for estrogen receptors, showcasing its value in selective ligand identification (Rollinger et al., 200 These instances highlight the critical role of virtual screening (VS) in propelling drug discovery forward, particularly through the use of robust datasets and advanced screening methodologies."(90)
8.2 Molecular Dynamics Simulations
Molecular dynamics (MD) simulations have been widely applied to investigate biomacromolecules such as proteins and nucleic acids. Advances in this field now enable researchers to simulate entire cells, offering deeper insights into the fundamental molecular mechanisms of life. These simulations are instrumental in exploring structural changes under varying conditions, analyzing drug interactions with biological targets, and characterizing protein behavior (Heidari et al., 2016b). Moreover, MD techniques allow the observation of rapid molecular events at atomic-level detail within sub-millisecond timescales in many biologically relevant systems (Borhani & Shaw, 2012) (91)
8.2 Table: 1 Computational Methods in Virtual Screening (92)
|
|
|
9. Challenges and limitations :
A large volume of data is essential for training AI systems effectively, making data accessibility a key factor in their success. However, obtaining information from multiple database providers can increase costs for companies seeking accurate predictive outcomes. In addition, the data used must be reliable and of high quality. Other challenges hindering the widespread adoption of AI in the pharmaceutical industry include a shortage of skilled professionals capable of managing AI-driven platforms, limited financial resources—particularly among smaller firms—concerns over potential job displacement, doubts about the reliability of AI-generated results, and the “black box” issue, referring to the lack of transparency in how AI systems reach their conclusion ( 93)
Although some pharmaceutical companies have already adopted artificial intelligence (AI), the industry was expected to generate approximately US$2.199 billion in revenue from AI-driven solutions by 2022. Between 2013 and 2018, the sector invested more than US$7.2 billion across over 300 deals. To maximize the benefits of AI, pharmaceutical firms must clearly communicate the realistic objectives and problem-solving capabilities that AI can offer. Cultivating a workforce of software engineers and data scientists who possess both robust AI expertise and a deep understanding of the company’s research and development priorities, as well as its commercial goals, is essential for leveraging AI platforms effectively (Research & Markets, 2019). (94)
Through advances in total synthesis, semi-synthetic methods, and biosynthetic engineering, scientists are actively exploring new classes of natural products (NPs) with antimicrobial potential, while also enhancing and refining existing ones. Moreover, antivirulence strategies present an alternative approach to infection control, with NPs that interfere with bacterial quorum sensing emerging as promising tools in this context (Atanasov et al., 2021; Merit et al., 20 (95)
One of the key hurdles in applying AI to the development of natural product-based medications is maintaining high data quality. The effectiveness of machine learning (ML) and deep learning models heavily relies on the accuracy and uniformity of the training data. Variations in format, precision, and measurement standards across different data sources can pose significant challenges. Incomplete datasets may introduce bias into models, necessitating the use of imputation methods to address missing information. To ensure that AI models deliver reliable predictions and can be generalized across different scenarios, rigorous validation is essential. Techniques such as cross-validation, which assess model performance across multiple data subsets, are instrumental in identifying issues like overfitting or underfitting (Saldívar González et al., 2022). (96)
Integrating artificial intelligence into drug research presents notable regulatory hurdles. It is essential for regulatory authorities to grasp the reasoning behind AI-driven decisions. To facilitate this, AI models must be designed with interpretability in mind, and the entire development process should be meticulously documented (Okibe & Samuel, 2024).
The effectiveness of AI in drug discovery can be significantly enhanced through its integration with omics technologies such as genomics, proteomics, and metabolomics. This synergy enables a deeper comprehension of biological systems and drug interactions. By merging data across various omics layers, researchers can achieve a holistic view of disease pathways and therapeutic mechanisms. AI plays a pivotal role in constructing and analyzing biological networks, pinpointing critical pathways, and predicting how alterations in these pathways might influence disease progression and treatment outcomes (Egwuatu et al., 2024; Paul et al., 2021; Ekpan et al., 2024). (97)
10. LIMITATION
While the review offers valuable insights, it is not without limitations. To begin with, the diversity in experimental models and cancer types investigated makes direct comparisons challenging. Additionally, the majority of the included studies are preclinical, leading to a limited number of clinical trials that assess the therapeutic efficacy of Peganum harmala and Nigella sativa compounds, especially within frameworks involving artificial intelligence. Furthermore, several studies lacked detailed methodologies and reproducibility information, increasing the risk of publication and selection biase(98)
While phytochemicals offer numerous benefits in drug development, several limitations can hinder their effectiveness, safety, and feasibility. One major challenge is the intricate and diverse nature of phytochemical profiles found in plants. This complexity often results in difficulties with consistent identification and extraction of specific compounds (99)
Environmental conditions—including climate, geographic region, and soil characteristics—significantly influence a plant's phytochemical composition, which poses challenges for achieving consistent standardizatio (100)
"Finally, potential safety and toxicity issues represent a significant constraint, as certain phytochemicals may elicit harmful effects. Therefore, thorough toxicological assessments are essential to ensure their safe application."(101)
11.Future pespectives
To fully harness the capabilities of AI-assisted phytochemical therapy, a number of strategic initiatives are essential.
To successfully bridge the gap between laboratory discoveries and clinical implementation, collaborative efforts are essential—bringing together oncologists, computational biologists, pharmacognosists, and AI specialists. This interdisciplinary approach fosters a seamless integration of traditional medical practices with cutting-edge therapeutic innovations (refer to Figure 6 and Table 3 for synergy
11. 1 . The convergence of artificial intelligence with natural product databases is set to evolve in transformative ways:
12.CONCLUSION:
Although challenges remain, the integration of AI into natural product research offers immense potential for advancing the development of therapeutic compounds. By harnessing cutting-edge innovations in artificial intelligence and machine learning, scientists are able to expand the frontiers of discovery, uncovering novel natural products with promising medicinal properties (104)
AI has revolutionized the development of herbal medicines by overcoming traditional limitations and streamlining key processes such as identifying bioactive compounds, verifying plant species, designing formulations, and predicting toxicity. This advancement has led to reduced costs, accelerated drug research, and enhanced reliability. When integrated into areas like pharmacovigilance, quality control, and synergy modeling, AI ensures that natural remedies remain consistently effective and trustworthy. Additionally, real-time technologies and deep learning-powered mobile applications have empowered non-specialists to participate in quality assurance and detect adulteration with greater ease (105)
Harnessing phytochemicals for drug discovery presents a compelling strategy for developing innovative therapeutic agents. When integrated with modern technologies, these natural compounds can be effectively utilized in the drug development pipeline. For instance, advancements in high-throughput screening and computational methods can greatly accelerate the identification and optimization of phytochemical-based treatments, enhancing the efficiency of therapeutic discovery (106)
REFERENCES


Mayuri Mohondkar*, Shivprasad Dhage, Dr. Sonali Uppalwar, Artificial Intelligence in Phytochemical Screening: Emerging Trends and Future Directions, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 11, 3270-3297 https://doi.org/10.5281/zenodo.17671254
 10.5281/zenodo.17671254
10.5281/zenodo.17671254
