Shivajirao S. Jondhale college of Pharmacy, Thane, India
Drug discovery has undergone a remarkable transformation over the past few decades, driven by advances in technology, molecular biology, and computational sciences. Traditional methods, which relied heavily on trial-and-error and high-throughput screening, are now being complemented and enhanced by innovative approaches such as artificial intelligence (AI), machine learning (ML), genomics, and structure-based drug design. These modern strategies have significantly accelerated the identification of potential drug candidates, improved target validation, and reduced the overall cost and time of development. Moreover, advances in personalized medicine and the integration of omics technologies have enabled a deeper understanding of disease mechanisms, leading to the discovery of more effective and safer therapeutic agents. This review highlights the recent breakthroughs, emerging technologies, and future trends shaping the field of drug discovery, emphasizing their collective role in transforming pharmaceutical research and improving patient outcomes.
The history of drug discovery begins in the early, the earliest periods of human civilization. In those prehistoric days, therapies were frequently found by accident or as a result of nature observation, usually, though not always, utilizing components taken from animals or plants, and not only as a physical cure but also for the purpose of spiritual healing. This trial-and-error method developed over the centuries, resulting in more methodical research techniques and progress. Modern drug discovery now relies heavily on scientific techniques, including molecular biology and advanced computing, to identify and synthesize new therapeutic agents.[1]
During the drug design and discovery process, possible novel therapeutic agents are determined using various clinical, experimental, and computational models.[2] Developments in organic chemistry during the 19th and early 20th centuries allowed researchers to create compounds, alter the structures of natural products, and ultimately create entirely synthetic molecules.[3] New drug development is an expensive, time-consuming, and failure-prone process.[4] The advent of a wide range of software and computational tools has greatly facilitated the process of drug research and development. Computer applications for online screening. In drug design and discovery, lead optimization, ligand-based design, and structure are commonly employed5.In medicinal chemistry, drug discovery is essential since it forms the basis for creating novel therapies to treat a variety of illnesses.[5]
Computational techniques are being increasingly investigated, applied, and valued in the areas of drug design, discovery, and production.[6] Introducing a new drug to the market is a highly challenging, risky, and costly endeavor concerning labor, finances, and time.[7]
Modern advances in drug discovery:
Computer aided drug discovery
What is Computer Aided Drug Discovery?
Computer Aided Drug Discovery (CADD) is the use of computational tools, software, and modeling techniques to design, analyze, and identify new drug candidates. It helps predict how drugs interact with biological targets, making the drug discovery process faster, cheaper, and more accurate.
Wu Z, Chen S, et al discovered that CADD encompasses all computer-aided methods utilized for the discovery, design, and enhancement of compounds that possess specific structures and characteristics. It involves several processes, such as identifying targets, virtually screening chemical libraries, optimizing lead compounds, and assessing potential toxicity, all of which are conducted through computational simulations.[8] Yang, W., et.al found that CADD is a discipline that merges computer science with biochemistry to speed up the process of drug discovery and development.[9]
The integration of rational drug design with structural biology results in the identification of new therapeutic agents. To achieve this goal, the Computer Aided Drug Design (CADD) center collaborates with structural biologists, biophysicists, and computational scientists to discover new chemical entities. Bisht, N., et al. (year) investigated and found that the utilization of CADD and bioinformatics tools offers advantages such as reduced costs, quicker time to market, deeper understanding of interactions between drugs and receptors, and an accelerated drug discovery and development process.[10]
Niazi, S. K., and Mariam, Z. (year) discovered that the emergence of CADD was supported by two key developments: the advancing field of structural biology that revealed the three-dimensional structures of biomolecules, and the significant increase in computational capabilities that allowed for complex simulations to be conducted in much shorter durations. CADD can be divided into two primary types: structure based CADD and ligand based CADD.[11]
Structure Based CADD
Structure-based CADD depends on the information of the target protein structure to calculate interaction energies for all compounds tried. It is for the most part favored where high-resolution basic information of the target protein are accessible, i.e., for solvent proteins that can promptly be crystallized. The central objective of structure-based CADD is to plan compounds that bind firmly to the target, i.e., with huge decrease in free energy, progressed DMPK/ADMET properties, and are target particular, i.e., have decreased off-target impacts as discovered by Sliwoski et al.[12]
Ligands based CDDS
Ligand-based medicate plan strategies are valuable within the absence of an exploratory 3D structure. Ligand-based medicate plan is a backhanded approach to encourage the improvement of pharmacologically dynamic compounds by considering particles that are associated with the natural target of interest.Due to the need of an experimental structure, the known ligand atoms that tie to the drug target are examined to get it the auxiliary and physicochemical properties of the ligands that relate with the specified pharmacological action of those ligands as investigated by Acharya et al.[13]
Molecular docking :
Morris, G. M. et al. discovered that molecular docking is a computational method used to predict how a ligand binds to a biological target, such as a protein or nucleic acid. It aims to determine the most stable ligand–receptor complex based on shape and chemical complementarity. The process involves two key steps: generating possible binding orientations and evaluating them using scoring functions to estimate binding affinity.[14]
There are two main types of molecular docking: rigid docking and flexible docking.
Rigid Docking:
In this method, both the ligand and receptor are considered fixed, without any movement or flexibility.
It is faster and easier to perform but may not fully capture real molecular movements during binding. Rigid docking works well when both the ligand and binding site remain mostly unchanged. Early tools like DOCK used this approach as developed by Kuntz, I. D. et al.[15]
Flexible Docking:
Trott, O. et al. developed the concept of flexible docking, in which ligands and sometimes the receptor can change shape during binding. This makes the simulation more realistic. The ligand’s flexibility is handled by testing different shapes, while the receptor’s movement can be modeled using methods like side-chain adjustments or induced fit. Modern tools such as AutoDock and FlexX use this approach to give more accurate binding results.[16]
Artificial intelligence in drug discovery
What is artificial intelligence?
AI in drug discovery and development uses intelligent algorithms to identify drug targets, design molecules, and predict safety and efficacy, speeding up and improving the process.
Bassey, G. E. et al. investigated how the time between discovering a new drug and developing it is getting longer because finding effective chemical compounds is becoming more difficult and time-consuming. To speed up this process, scientists in medicinal chemistry are now using artificial intelligence (AI) to improve and modernize the pharmaceutical industry. The traditional approach to drug discovery is a complex and time-intensive process that depends heavily on manual efforts, including extensive screening and experimental trial-and-error methods.[17] Ocana, Alberto, et al. Found that traditional methods in drug discovery and early clinical development have worked well and helped create new medicines, but AI can make these methods even better. It can also bring new ideas that make the whole process faster and more efficient.[18]
Blanco-González, A. et al. investigated how collaboration between AI researchers and pharmaceutical scientists plays a vital role in developing innovative and effective treatments. By merging their expertise, they can design advanced algorithms and machine learning models to predict drug efficacy and accelerate the discovery process. AI also enhances the accuracy of clinical trials by analyzing large datasets to identify patterns, potential side effects, and optimal drug candidates. This partnership enables pharmaceutical companies to make faster, data-driven decisions. Moreover, AI-assisted analysis of population data supports personalized medicine and cost-effective healthcare. A notable example is the collaboration between Merck and Numerate to apply AI in medicinal chemistry.[19]
Tabe no. 1 Difference in classical and ai driven drug development
|
Classical drug development |
Ai driven drug development |
|
Target Discovery: Illustrate visuals of gene/protein interactions or omics data |
Target Prediction (QSAR): Al models predicting target interactions |
|
Compound Identification: Show compound libraries or molecular structures |
Chemical Structure Design: Al models create novel molecular structures or designs |
|
In Vitro Preclinical Evaluation: Depict cell culture plates or laboratory apparatus |
Molecular Docking: Depict simulations of protein-ligand docking |
|
Mode of Action: Assessment use proteomics or protein interaction visual illustrations |
Molecular Dynamics (MD):Utilize MD conformational or structural simulation images |
Challenge and limitations of using AI in drug discovery :
Vamathevan, J. et al. investigated that although AI offers many potential advantages in drug discovery, several challenges and limitations still need to be addressed. A major issue among these is the limited availability of appropriate and high-quality data. [20]Tsuji, S. et al. Discovered that AI-driven methods generally depend on vast amounts of data for effective training.[21] Gómez-Bombarelli, R. et al. Found that in many situations, researchers may face challenges due to limited access to sufficient data. Even when data is available, it might be incomplete, inconsistent, or of low quality. Such issues can make it difficult for artificial intelligence systems to learn effectively and produce dependable predictions. As a result, the accuracy, reliability, and overall performance of the outcomes can be significantly affected, leading to potential errors or misleading conclusions in the drug discovery process.[22]
Techniques in artificial intelligence:
Deep learning:
Sarker, I. H. explained that deep learning (DL) is a part of machine learning (ML) and artificial intelligence (AI). It is now seen as an important technology of the Fourth Industrial Revolution (Industry 4.0). Because it can learn from data, deep learning, which is based on artificial neural networks (ANN), has become very popular in computer science. It is used in many fields such as healthcare, image recognition, text analysis, cybersecurity, and others.[23] Hao, Z. Investigated that it uses several layers of processing to learn and understand complex information from data through advanced structures and transformations.[24] Sarker, I. H. et al. Found that Deep learning is a smaller part of machine learning and also belongs to the larger field of artificial intelligence, which aims to make machines think and act like humans,[25] Machine learning (ML) is a technique that enables systems to gain knowledge and improve performance based on data or past experiences,[26] Deep learning (DL) involves techniques that learn from data using multiple layers of neural networks to perform computations and data processing.[23]
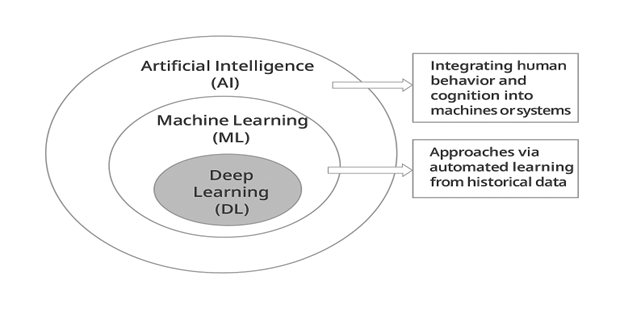
Figure 1 “Hierarchy of Artificial Intelligence (AI), Machine Learning (ML), and Deep Learning (DL)”
Reinforcement learning:
Bou, A. et al.found that reinforcement learning (RL) is now seen as a useful method to quickly and effectively explore the large chemical space.RL is a group of machine learning methods that use feedback as a signal to improve and guide the decision-making process.Thus, RL algorithms can adjust the molecule-building decision process to obtain specific molecular features, leading to the creation of new molecules with desired properties. This serves as a strategy for exploring the chemical space. RL depends on a reward function that evaluates each molecule based on the particular application or research goal. The RL algorithm aims to maximize this value, guiding the decision-making and exploration of chemical space—especially useful when the space is too large or complex to be fully searched or scored.[27]
Fragment-Based Drug Design:
What is Fragment based drug design?
Fragment-Based Drug Design (FBDD) is a method of drug discovery where small chemical fragments, which bind weakly to a biological target, are identified and then optimized or combined to develop potent drug molecules.
AlKharboush, D. F. et al. Investigated that Fragment-based drug discovery (FBDD) has emerged as a key approach in modern drug development, enabling the efficient identification and optimization of novel lead compounds with high chemical diversity.[28] Li, Q. Found that FBDD starts with small chemical fragments that have simple structures, low molecular weight (under 300 Da), and weak binding to the target.[29] Erlanson, D. A., & Jahnke, W. investigated that Since it was first introduced in 1981, fragment-based drug design has shown to be a useful and promising method for finding potential hit molecules in the early stages of drug discovery.[30] Fragment-based drug discovery focuses on screening small, low molecular weight compounds that show weak initial binding to target proteins but can be refined into potent drugs through medicinal chemistry. Shuker, S. B. discovered In the 1990s, the “SAR by NMR” method was introduced, demonstrating how NMR spectroscopy can be used to detect and study small fragment binders.[31] Today, FBDD has evolved into a widely adopted screening strategy employed across pharmaceutical industries, biotechnology firms, and academic research centers.
Bon, M. investigated that Fragment libraries can explore a significantly larger portion of chemical space than HTS libraries while using far fewer compounds. Unlike complex molecules, which often cause unfavorable interactions or steric clashes with the target, fragments tend to form more efficient and optimal atom-level binding interactions.[32] Therefore, Murray, C. et.al found that even a library containing just one to two thousand small molecules can effectively yield valuable hits for a drug discovery program.[33] Furthermore,Kirsch, P. Discovered that fragment hit rates can serve as an indicator of a target’s druggability and help in identifying challenging binding regions, including allosteric sites or small “hot spot” binding pockets commonly involved in protein–protein interactions.[34] Finally, Murray, C. W., & Rees, D. C. Investigated that the approach allows exploration of a broader chemical space using fewer compounds, increasing the chances of finding novel scaffolds.[35]
Jhoti, H. et al. discovered that biophysical techniques such as nuclear magnetic resonance (NMR), X-ray crystallography, and surface plasmon resonance (SPR) are commonly used to detect fragment binding and determine structural interactions.[36] Once fragment hits are identified, Erlanson, D. A. found that they can be grown, linked, or merged to create higher-affinity molecules with desirable pharmacokinetic properties.[37] In addition, Chen, H. investigated combining FBDD with computational methods such as molecular docking, machine learning, and artificial intelligence has further enhanced fragment screening efficiency and binding prediction accuracy.[38]
Overall, Al Kharboush, D. F. concluded that FBDD represents a powerful, efficient, and cost-effective approach for discovering novel drug candidates with improved selectivity, efficacy, and delivery properties.[39]
De novo drug discovery:
What is de novo drug discovery?
De novo drug discovery is a method of designing new drug molecules from scratch using computational tools, based on the structure of a biological target, rather than modifying existing compounds.
Atz, K. et al. Investigated that De novo drug discovery means making new drug molecules completely from scratch, not from old drugs.[40] Crucitti, D. et al. investigated that it uses computer tools, chemistry, and artificial intelligence (AI) to design new medicines.[41] Mouchlis, V. D. et al. found that older methods used known chemical parts, so they could only make small changes to existing drugs.[42] Schneider, G. discovered that, now AI-based programs can explore many more new and creative chemical ideas.[43]
Modern Computer and AI Tools:
Tang, Y. and Moretti, R. discovered that ,New deep learning models like VAEs and GANs can create new molecules that look like real drugs.[44] Ang, D. investigated that Other AI tools like Transformers and diffusion models make safe and useful molecules faster.[45] Gao, W. et al. found that Reinforcement learning helps the computer improve designs step by step to make better drugs.[46]
Using Structures and Experiments Together:
Jiménez-Luna, J. et al. investigated that AI-based scoring systems and docking programs help test which molecules might work best.[47] Lin, Y. et al. discovered that tools like AlphaFold help design drugs that fit well into their protein targets.[48] Atz, K. et al. found robotic machines can quickly make and test these molecules, saving time in experiments.[49] Brown, N. et al. Found that deep learning also helps predict how strongly a molecule will bind to its target protein.[50]
Challenges and Future Possibilities:
Zhavoronkov, A. et al. found that there are still problems like AI models being hard to understand and errors in data.[51] Ouma, R. B. O. et al.suggested that new technologies such as quantum computing and safe data sharing (federated learning) may help fix these issues. [52] Gao, K. et al. Investigated that in the future, AI-made drugs could become real medicines if they pass proper clinical testing.[53]
High-Throughput and High-Content Screening:
What is HTS and HCS ?
High Throughput Screening (HTS) is a technique that rapidly tests large numbers of compounds for biological activity using automated systems. High Content Screening (HCS) goes a step further by not only testing compounds but also analyzing detailed cellular responses (like morphology, protein expression, or signaling) using imaging and quantitative data.
Inglese, J. et al. discovered that High-throughput screening (HTS) and high-content screening (HCS) have become fundamental tools in modern drug discovery, enabling scientists to assess large chemical libraries for biological activity within a short time frame.[54] Pushpakom, S. Investigated that these screening platforms combine automation, robotics, and advanced data analytics to identify potential therapeutic compounds efficiently.[55] Macarron, R. Found that HTS focuses on testing compound activity against specific targets using biochemical or cell-based assays, while HCS provides detailed phenotypic information using imaging technologies.[56] Mayr, L. M. and Bojanic, D. concluded that together,these strategies accelerate early-stage drug discovery by reducing experimental time, cost, and human error.[57]
Tchniques in HTC and HCS:
Automated Robotic Screening
Segler, M. H. S. et al. Found that automation lies at the heart of HTS, involving robotic liquid handling, automated plate readers, and integrated data systems that minimize human intervention.[58] Ghosh, A. et.al. discovered that Robots can precisely pipette small reagent volumes into microtiter plates, perform incubation, and detect activity using fluorescence, luminescence, or absorbance-based assays.[59] Wang, D. et al. Found that Automation improves reproducibility and throughput while reducing contamination risk.[60] Wu, H. et al. investigated that modern robotic platforms are now AI-assisted, optimizing assay conditions in real-time to improve data accuracy.[61] Additionally, Jiang, Y. et al. discovered that machine learning models predict compound interactions and reduce the need for redundant screening, further enhancing productivity.[62] Li, X. et al. concluded that automated screening systems are widely used in pharmaceutical companies for target validation, compound library screening, and secondary assay confirmation.[63]
Microarray-Based Assays :
Boutros, M., and Heigwer, F. Investigated that Microarray assays provide another layer of high-throughput capability by immobilizing thousands of biological probes—such as DNA, RNA, peptides, or proteins—onto a single solid surface.[64] Caicedo, J. C. et al. investigated that these assays enable simultaneous detection of interactions between small molecules and biomolecular targets, making them crucial for understanding disease mechanisms.[65] Bray, M.-A. et al. discovered that protein and antibody microarrays have been widely applied for identifying binding partners and signaling pathways in oncology and immunology.[66] Singh, S. discovered that the combination of microarrays with nanotechnology and microfluidics has improved detection accuracy and signal amplification. Additionally, integration with microfluidic devices allows rapid reaction times and minimal reagent consumption.[67]
Cell-Based Imaging Systems:
Way, G. P. et al. investigated that cell-based imaging systems are essential components of high-content screening platforms that enable quantitative visualization of cellular responses under various conditions.[68] Caicedo, J. C. et al. discovered that these systems combine automated microscopy, fluorescence labeling, and computerized image analysis to capture complex phenotypic information.[69] Boutros, M. et al. found that both live-cell and fixed-cell imaging modes are used to observe dynamic processes like mitosis, apoptosis, or intracellular trafficking.[70] Typically, cells are seeded in multiwell plates (96-, 384-, or 1536-well), treated with chemical or genetic perturbations, stained with fluorescent dyes, and imaged automatically. Bray, M.-A. et al. Investigated that high-throughput robotic microscopes capture hundreds of fields per well, ensuring statistically reliable datasets for downstream analysis.[71] Chandrasekaran, S. N. et al. investigated automated segmentation software measures multiple cellular features—shape, texture, intensity, and localization—producing rich phenotypic profiles.[72] Ghosh, A. et al. discovered that Machine learning improves accuracy and scalability, making analysis faster and more reproducible.[73] Qian, X. et al. found that recent advances like 3D organoid imaging and multiplex fluorescence microscopy enhance physiological relevance and depth of information.[74] Way, G. P. et al. emphasized that by combining automation, artificial intelligence, and microscopy, these systems have transformed modern biomedical research and accelerated drug development.[75]
Advanced Preclinical Models:
Ingber, D. E. discovered that advanced preclinical models are new scientific tools that help researchers test drugs in ways that are closer to how the human body actually works.[76] Low, L. A., et al. found that they improve how well lab results match real patient outcomes and also reduce the need for testing on animals.[77]
Organ-on-chip (OOC) :
An organ-on-a-chip (OoC) is a small device made with micro-sized channels that contain living human cells as investigated by Leung, C. M.[78] It helps scientists create conditions similar to real organs, such as blood flow, pressure, and nutrients, as discovered by Morais, A. S[79]. van der Helm, M. W. found that using this method, small models of the heart, liver, and lungs can be made to test how medicines work or cause side effects.[80] However, Zhang, B. emphasized that although OOC is very useful, it is still difficult to make chips for many organs at once or use them on a large scale.[81]
3D Bioprinting:
Mirsky, N. A. et al. investigated that 3D bioprinting is a new technology that builds living tissues layer by layer using special materials called bioinks, which contain living cells.[82] Sachdev, A. et al. discovered that this process can make tissues that look and act like real organs and are useful for testing drugs safely before human trials.[83] Brandt, A. et al.found that Scientists also use bioprinting to make patient-specific tissues for personalized treatments or to practice surgeries.[84] Qu, S. et al. emphasized that, However, problems like keeping the cells alive, creating small blood vessels, and making strong tissue structures still need to be solved.[85]
Patient-Derived Organoids (PDOs):
Brandt, A. et al. investigated that patient-derived organoids (PDOs) are tiny 3D versions of organs made from a patient’s own cells.[84] Qu, S. et al. discovered that these organoids keep many of the same genes and features as the original tissue, making them very helpful for studying how diseases grow and how patients respond to drugs.[85] Tong, L. et al. found that PDOs are also used to make collections (called biobanks) for testing many medicines on different types of patient samples.[86] Kim, J. et al. Discovered that the scientists are still working on adding immune cells and improving the environment inside these organoids to make them even more realistic.[87].
AI–Pharma Collaborations and Cloud Computing:
Paul, D., and Sanap, G. investigated how Artificial intelligence (AI) and cloud computing are transforming pharmaceutical research by integrating massive datasets and computational power to accelerate drug discovery.[88] Serrano, D. R. et al. discovered that these technologies allow researchers to analyze biological data, simulate molecular interactions, and identify novel therapeutic compounds more efficiently.[89] Su, J. et al. found that Cloud platforms enable scalability, global collaboration, and data storage for AI-driven pharmaceutical innovation.[90] Zhavoronkov, A. et al. Investigated that collaborations between AI technology firms and pharmaceutical industries are essential to improving drug discovery pipelines and reducing development timelines.[91] Bess, A. et al. Found that by merging biological insights with advanced algorithms, these partnerships enhance both prediction accuracy and experiment reproducibility.[92]
Example’s:
DeepMind’s AlphaFold:
Jumper, J., and Evans, R. investigated deepMind’s AlphaFold represents a major breakthrough in structural biology by accurately predicting 3D protein structures from amino acid sequences.[93] Senior, A. W. et al. discovered that, this AI model outperformed experimental methods in accuracy and speed, solving one of biology’s grand challenges.[94] Varadi, M. found that, Its database, the AlphaFold Protein Structure Database, provides millions of freely available predicted structures, aiding scientists globally in drug target identification.[95] Skolnick, J., and Gao, M. Investigated that researchers now use AlphaFold’s data for structure-based drug design, particularly in oncology and infectious diseases.[96]
Insilico Medicine:
Zhavoronkov, A. et al. investigated how insilico Medicine integrates deep learning, reinforcement learning, and cloud computing to generate new drug-like molecules in record time.[97]
Zhavoronkov, A. et al. discovered that Insilico Medicine is a leading company that uses artificial intelligence (AI) to design and discover new drug molecules quickly and efficiently.[98] Zhavoronkov, A. et al. Investigated that During the COVID-19 pandemic, the company demonstrated its generative AI approach by identifying potential antiviral compounds targeting the SARS-CoV-2 protease.[99] Serrano, D. et al. found that Its AI-based tools such as Chemistry42 and PandaOmics combine molecular modeling, deep learning, and bioinformatics to improve early drug discovery.[100] Vamathevan, J. et al. Found that AI-driven discovery, like Insilico’s work, can help reduce the cost and time required to find effective drug candidates.[101] However, despite these advances, experts note that AI-discovered compounds still require full clinical validation before entering the market. Overall, Lavecchia, A. et al. concluded that Insilico Medicine represents one of the best examples of how AI can reshape modern drug discovery, but real-world results will depend on successful clinical testing.[102]
BenevolentAI and Atomwise:
Hopkins, A. L. et al. investigated BenevolentAI and Atomwise are two leading companies applying artificial intelligence (AI) in drug discovery. BenevolentAI uses machine learning and knowledge graphs to identify novel drug targets and repurpose existing drugs efficiently, accelerating the development process.[103] Chen, H. et al. found that atomwise, on the other hand, employs deep learning–based molecular structure analysis to predict binding affinity between small molecules and proteins, enabling faster virtual screening and compound selection.[104] Schneider, G. et al. reported that Both companies demonstrate how AI can reduce time and cost in early-stage drug discovery, offering more accurate predictions than traditional methods.[105]
Quantitative Structure–Activity Relationship (QSAR):
Perkins, R. et al. investigated QSAR is a computational approach that relates a molecule’s chemical structure to its biological or physicochemical activity using mathematical models.[106] Bastikar, V. et al. discovered that It helps predict the activity of new compounds without laboratory testing and is widely used in drug discovery and environmental toxicology.[107]
2D QSAR:
Myint, K. Z. et al. investigated 2D-QSAR uses molecular descriptors such as molecular weight, logP, and topological indices derived from 2D chemical structures.[108] Hansch, C. et al. discovered that these models often employ statistical or machine learning techniques like multiple linear regression or support vector machines.[109] Gramatica, P. et al. found that 2D-QSAR is simple, fast, and effective for large datasets, but it cannot describe 3D conformational effects of molecules.[110]
3D QSAR:
Jagiello, K. et al. investigated 3D-QSAR adds spatial information, considering steric and electrostatic fields to better understand molecular interactions.[111] Cramer, R. D. et al. discovered that common 3D methods such as CoMFA and CoMSIA provide visual maps of active sites and structure–activity trends.[112] Srour, A. M. et al. found that these models require accurate molecular alignment and are computationally intensive .[113]
current trends:
Vedani, A. et al. investigated recent developments including fragment-based, 4D-, and 5D-QSAR approaches that integrate time and solvent effects.[114] Muratov, E. N. et al. discovered that machine learning and deep learning have further enhanced QSAR accuracy and generalization power.[115] Soares, T. A. et al. found that overall, QSAR continues to evolve as a vital computational tool in cheminformatics and molecular modeling. It is now a vital tool for designing drugs, predicting toxicity, and reducing animal testing in chemical safety assessment.[116]
FUTURE PROSPECTS:
The future of drug discovery and development is expected to be increasingly driven by the integration of artificial intelligence (AI), machine learning (ML), and advanced computational approaches, which can efficiently analyze large biological and chemical datasets to identify novel drug targets, design new molecules, and predict pharmacokinetic and toxicity profiles at early stages.[117] These technologies will significantly reduce the time, cost, and failure rates traditionally associated with drug development. Alongside computational advances, automation, robotics, and high-throughput screening (HTS) platforms will further enhance experimental efficiency and reproducibility, enabling rapid evaluation of thousands of compounds with minimal human intervention.[118] In addition, the scope of drug discovery will expand beyond conventional small molecules to include novel therapeutic modalities such as peptide drugs, oligonucleotides, gene and cell therapies, antibody–drug conjugates, and targeted protein degradation technologies like PROTACs. These approaches offer new strategies to treat complex diseases and target proteins previously considered undruggable.[119] The growing use of genomics, proteomics, and other omics technologies will support precision and personalized medicine by allowing drugs to be tailored to specific patient populations based on molecular and genetic profiles.[120] Furthermore, advances in silico ADME-Tox modeling and systems biology approaches will improve prediction of drug behavior in humans, reducing late-stage clinical failures and dependence on animal testing.[121] Overall, the convergence of AI-driven computation, innovative drug modalities, and data-driven biological understanding will make future drug discovery more efficient, precise, and patient-centric.
SUMMARY:
Drug discovery has significantly advanced over the past few decades due to the integration of modern science and technology. Earlier, drug discovery mainly relied on trial-and-error methods and extensive laboratory testing, which were time-consuming, expensive, and had high failure rates. Today, this process is supported by advanced computational, biological, and data-driven approaches that improve efficiency and accuracy.
Computer-Aided Drug Discovery (CADD) plays a central role by using computational tools to predict how drug molecules interact with biological targets. Techniques such as molecular docking, molecular dynamics, and Quantitative Structure–Activity Relationship (QSAR) help in understanding drug–target interactions and optimizing lead compounds. Fragment-based drug design and de novo drug design further enable the creation of novel drug molecules with improved potency and selectivity.
Artificial intelligence (AI) and machine learning have transformed drug discovery by analyzing large biological datasets, predicting drug efficacy and toxicity, and accelerating target identification. High-throughput screening (HTS) and high-content screening (HCS) allow rapid testing of thousands of compounds, reducing time and experimental cost. Moreover, advanced preclinical models such as organ-on-chip systems, 3D bioprinting, and patient-derived organoids better mimic human physiology, improving the prediction of clinical outcomes.
Overall, these advances have made drug discovery faster, more cost-effective, and more precise. The integration of biology, chemistry, computation, and AI is driving the development of safer, more effective, and personalized medicines for complex diseases.
CONCLUSION
Recent advances in drug discovery have revolutionized the way new therapeutics are identified, designed, and developed. The integration of computational modeling, artificial intelligence, omics technologies, and high-throughput screening has significantly accelerated the process while improving accuracy and reducing costs. Modern approaches such as molecular dynamics, structure-based drug design, and machine learning have enabled researchers to predict drug–target interactions with unprecedented precision. Moreover, the rise of personalized and precision medicine is transforming drug discovery into a more patient-centered science, offering treatments tailored to individual genetic and molecular profiles. Despite ongoing challenges in validation and clinical translation, these innovations collectively represent a paradigm shift toward faster, smarter, and more effective drug development. The future of drug discovery thus lies in the seamless fusion of biology, chemistry, data science, and technology to address complex diseases and unmet medical needs.
REFERENCES







Purva Gambhirrao, Ghodgaonkar S., Jyoti Sonawane, Chaitali Gaikar, Pragati Gharatkar, Sanchita Ghode, Aditi Ghadge, Advances in Drug Discovery and Development, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 4114-4133. https://doi.org/10.5281/zenodo.19354480
 10.5281/zenodo.19354480
10.5281/zenodo.19354480
