Satara College Of Pharmacy, Satara
Spinal muscular atrophy (SMA), a rare genetic disorder causing progressive muscle weakness due to SMN1 gene mutations, affects approximately 1 in 10,000 live births and historically lacked disease-modifying treatments. The approval of Spinraza (nusinersen) with the aid of the FDA in 2016 marked a paradigm shift because the first therapy for SMA, leveraging antisense oligonucleotide (ASO) technology to decorate SMN protein production via SMN2 gene splicing. Developed by Ionis Pharmaceuticals and marketed by Biogen, Spinraza has demonstrated significant efficacy across SMA types, with clinical trials like ENDEAR and CHERISH showing motor milestone achievements and survival benefits, particularly with early intervention. Its safety profile is favorable, though intrathecal administration poses challenges. Regulatory incentives under the Orphan Drug Act expedited its approval, yet its high cost—$750,000 for the first year— raises access and ethical concerns. Compared to emerging therapies like Zolgensma (gene therapy) and Evrysdi (oral small molecule), Spinraza offers broad applicability but requires repeated dosing. Patient and caregiver experiences highlight improved quality of life alongside logistical burdens, while real-world data as of April 05, 2025, affirm long-term benefits. This review synthesizes Spinraza’s mechanism, clinical outcomes, pharmacokinetics, regulatory journey, economic implications, and future directions, positioning it as a cornerstone of precision medicine for rare diseases. Despite its transformative impact, equitable access remains a critical challenge, underscoring the need for policy innovation to maximize its global reach.
Spinal muscular atrophy (SMA) ranks among the most severe rare genetic disorders, characterized by progressive muscle wasting and weakness stemming from the degeneration of motor neurons in the spinal cord. With an incidence of approximately 1 in 10,000 live births worldwide [27], SMA has long epitomized the challenges of rare disease management within neuromuscular medicine. As an autosomal recessive condition, it arises primarily from mutations or deletions in the survival motor neuron 1 (SMN1) gene, resulting in insufficient production of the SMN protein—a vital component for motor neuron survival [18]. For decades, clinicians and families confronted a bleak prognosis, with supportive measures like ventilatory assistance and physical therapy as the sole interventions, often inadequate against SMA’s relentless progression [12]. This dire scenario transformed dramatically on December 23, 2016, when the U.S. Food and Drug Administration (FDA) approved Spinraza (nusinersen), marking the first disease- modifying therapy for SMA [9]. Developed by Ionis Pharmaceuticals, a pioneer in antisense oligonucleotide (ASO) technology, and commercialized by Biogen, Spinraza represents a pinnacle of precision medicine. This ASO targets the SMN2 gene—a near-identical paralog to SMN1— enhancing its capacity to produce functional SMN protein through a sophisticated mechanism of RNA splicing modulation [15]. Its approval not only offered hope to SMA patients but also underscored the potential of orphan drug development, a field bolstered by legislative incentives like the Orphan Drug Act of 1983, which supports treatments for rare diseases affecting fewer than 200,000 individuals in the U.S. [14]. Since its debut, Spinraza has reshaped clinical outcomes, sparked debates over cost and accessibility, and set a precedent for genetic therapies in rare diseases. This review provides an exhaustive analysis of Spinraza’s multifaceted impact. It explores the molecular basis of SMA, Spinraza’s development and mechanism, its clinical efficacy and safety, pharmacokinetic properties, regulatory milestones, economic challenges, patient and caregiver experiences, comparisons with emerging SMA therapies, and long-term implications as of April 05, 2025. By integrating scientific rigor, clinical evidence, and societal perspectives, this article aims to offer a definitive resource on Spinraza’s role in addressing one of medicine’s most pressing unmet needs.
Pharmacokinetics of Spinraza
Spinraza is administered intrathecally at 12 mg per injection directly into the CSF, bypassing the blood-brain barrier. After administration, it reaches peak CSF concentrations within 1–6 hours and exhibits a long CSF half-life of 135–177 days, allowing durable target engagement with infrequent dosing [11]. Systemic exposure is minimal (<1% plasma levels), it avoids cytochrome P450 metabolism, and is primarily degraded by exonucleases [3].
Pharmacodynamics of Spinraza
Pharmacodynamically, Spinraza binds to the ISS-N1 site in intron 7 of SMN2 pre-mRNA, blocking splicing repressors (hnRNP A1/A2) and facilitating exon 7 inclusion [15]. This increases full-length SMN protein production, restoring motor neuron function and halting neurodegeneration [2]. Clinical trials ENDEAR and CHERISH confirm motor milestone improvements and survival benefits [11, 21].
II. MATERIALS AND METHODS / METHODOLOGY
This comprehensive review was conducted through an extensive and structured analysis of peer-reviewed literature, clinical trial data, regulatory documents, and real-world evidence related to Spinraza (nusinersen) and spinal muscular atrophy (SMA). The objective was to critically evaluate the efficacy, safety, pharmacology, and socioeconomic impact of Spinraza from its development through its global implementation.
Data Sources:
Relevant scientific articles were retrieved from:
- PubMed
- Google Scholar
- ScienceDirect
- ClinicalTrials.gov
- FDA and EMA official portals
- Cure SMA Foundation reports
Search Strategy:
A keyword-based search was employed using combinations of the following terms:
"Spinraza", "nusinersen", "Spinal Muscular Atrophy", "ASO therapy", "SMN1", "SMN2", "clinical trials", "ENDEAR", "CHERISH", "SHINE", "orphan drugs", "gene therapy", "Zolgensma", "Evrysdi"
Search filters were applied to include articles published between 2015 and 2025 in English.
Inclusion Criteria:
- Peer-reviewed articles, clinical studies, and real-world observational data related to nusinersen (Spinraza)
- Publications discussing SMA classification, genetics, and diagnosis
- Reports from international health agencies and pharmaceutical regulatory authorities
Exclusion Criteria:
- Non-English publications
- Editorials or unverified anecdotal reports without data support
Data Extraction and Analysis:
Information was categorized into:
- Molecular basis of SMA
- Mechanism of action of Spinraza
- Clinical efficacy and safety outcomes
- Pharmacokinetic and pharmacodynamic profiles
- Regulatory milestones and approvals
- Cost, accessibility, and ethical considerations
- Comparative analysis with emerging therapies (Zolgensma and Evrysdi)
- Real-world patient experiences and long-term impact
Data from clinical trials (e.g., ENDEAR, CHERISH, SHINE) were critically reviewed, and statistical outcomes were cited directly from published sources. Additionally, patient registries and treatment guidelines were examined to assess the real-world applicability of Spinraza.
III. SPINAL MUSCULAR ATROPHY: A DETAILED DISEASE BACKGROUND
Spinal muscular atrophy (SMA) is a hereditary neuromuscular disorder driven by the loss of alpha motor neurons in the spinal cord’s anterior horn, leading to progressive muscle weakness, atrophy, and, in severe cases, respiratory failure. The root cause is mutations or deletions in the SMN1 gene on chromosome 5q13, which encodes the survival motor neuron (SMN) protein essential for RNA metabolism and motor neuron integrity [18]. SMA’s autosomal recessive nature requires two defective SMN1 alleles for disease manifestation, with carriers typically asymptomatic due to sufficient SMN production from one functional copy. SMA’s clinical presentation varies widely, categorized into five types based on age of onset and motor milestones [21]:
Table 1: Types Of SMA Based On Age Of Onset And Motor Milestones
|
Sma Type |
Onset |
Key Features |
Motor Functions |
Prognosis |
|
Type 0 |
Prenatal |
Severe Hypotonoia, Respiratory Failure At Birth. |
No Motor Milestones. |
Fatal Within Weeks Without Support. |
|
Type 1 |
< 6 Months |
Weakness, Can Not Sit, Feeding Issues. |
Can Not Sit Unaided. |
Death By Age 2 Without Treatment. |
|
Type 2 |
6-18 Months |
Can Sit But Can Not Walk, Scoliosis, Joint Issues. |
Sits, No Walking. |
Progressive With Respiratory Decline. |
|
Type 3 |
>18 Months |
Walks , Later Mobility Loss, Proximal Weakness. |
Walks Initialy. |
Slow Progression, Mobility Loss. |
|
Type 4 |
Adult Hood |
Mild Weakness, Fatigue. |
Normal Motor Milestones |
Very Slow Progression, Normal Life. |
The SMN2 gene, a near-identical backup, modifies severity. A C-to-T transition in exon 7 disrupts splicing, producing mostly truncated, unstable protein, though 10–15% of transcripts yield functional SMN [19]. SMN2 copy numbers (1–4 typically) inversely correlate with severity; Type 1 patients often have 1–2 copies, while Type 4 patients may have 4 or more [10]. This variability complicates prognosis and treatment planning, as copy number assays (e.g., qPCR) are now standard in diagnostics. Historically, SMA diagnosis lagged due to its rarity and symptom overlap with conditions like muscular dystrophy or congenital myopathies, relying on electromyography and muscle biopsies before genetic testing emerged in the 1990s [24]. Early cases were often misdiagnosed, delaying care. Today, newborn screening—adopted in 48 U.S. states and parts of Europe—detects SMN1 deletions via dried blood spots, enabling intervention within weeks of birth, a game-changer for therapies like Spinraza [24]. Understanding SMA’s genetic and clinical complexity is foundational to appreciating Spinraza’s targeted approach.
IV. THE ROLE OF ORPHAN DRUGS IN ADDRESSING RARE DISEASES
Orphan drugs target rare diseases, defined in the U.S. as affecting fewer than 200,000 individuals and in the EU as less than 5 in 10,000 [14]. SMA, with its 1 in 10,000 incidence, exemplifies this category [27]. Over 7,000 rare diseases impact 400 million people globally, yet fewer than 10% have approved treatments, highlighting a vast therapeutic void [22]. Small patient populations historically deterred pharmaceutical investment due to limited profitability, leaving conditions like SMA, Pompe disease, and cystic fibrosis underserved until policy interventions emerged. The U.S. Orphan Drug Act of 1983 and EU Regulation (EC) No 141/2000 transformed this landscape [8,7]. The U.S. law offers tax credits (up to 50% of clinical trial costs), research grants, fee waivers, and 7-year market exclusivity, while the EU provides 10-year exclusivity and protocol assistance [14] Since 1983, over 600 orphan drugs have been approved in the U.S.—versus 10 pre-Act—including treatments like imatinib for chronic myelogenous leukemia and enzyme replacements for Gaucher disease [14]. Spinraza’s development reflects this shift, navigating scientific hurdles (e.g., CNS delivery) and economic barriers (e.g., $1 billion RCD cost) to serve SMA’s small cohort [13]. Its success extends beyond SMA, inspiring ASO research for Huntington’s disease and spinal cerebellar ataxia, affirming orphan drugs’ broader societal impact [2].
V. SPINRAZA: DEVELOPMENT, MECHANISM, AND ADMINISTRATION
Spinraza’s creation is a landmark in RNA-targeted therapy. As an ASO, it comprises a synthetic 18-nucleotide strand with a phosphorothioate backbone, binding SMN2 pre- mRNA at intron 7 to block splicing repressors (e.g., hnRNP A1), promoting exon 7 inclusion and boosting functional SMN protein [15].
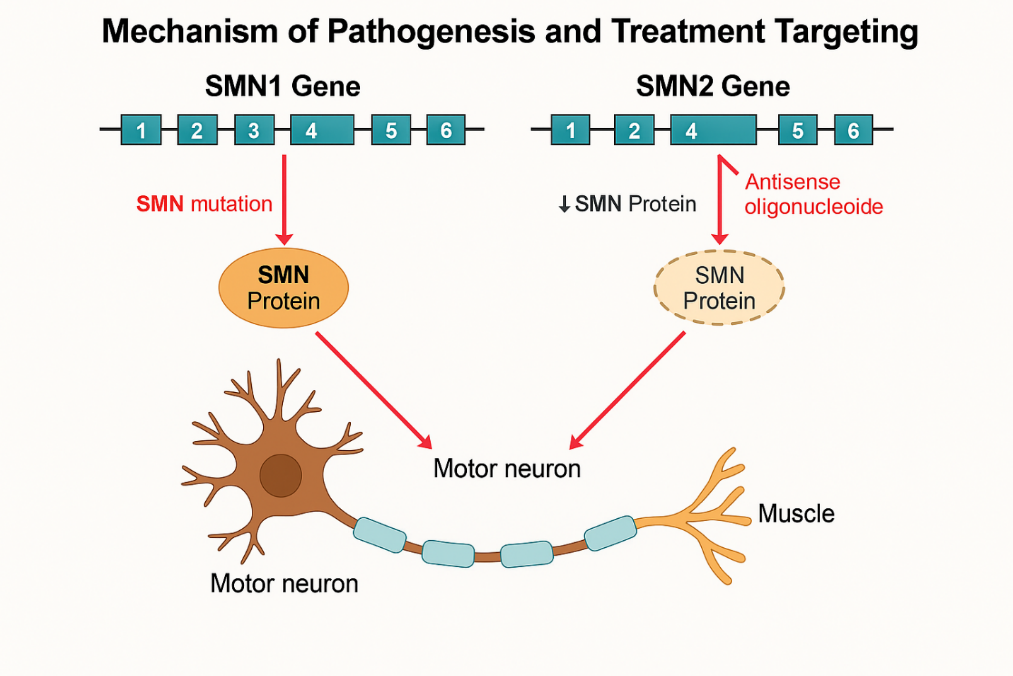
Figure 1: Mechanism Of Pathogenesis And Treatment Of SMA
Ionis Pharmaceuticals pioneered this approach, building on 1980s splicing research and 2000s preclinical studies in SMA mouse models, where ASOs increased SMN levels, extended survival from 10 to over 100 days, and restored motor function [25].
Table 2: Timelines And Milestones Of SMA.
|
Timelines |
Milestones |
|
1980s |
Splicing Mechanisms Discovered |
|
2000s |
Preclinical Aso Trials In Mice |
|
2010s |
Clinical Trials And Fda Approval |
|
2016s |
Clinical Trials And Fda Approval |
Clinical translation required optimizing stability, specificity, and delivery to the CNS, a decade- long effort involving over 50 scientists and multiple Phase 1 trials. Administered intrathecally into the cerebrospinal fluid at 12 mg per dose, Spinraza bypasses the blood-brain barrier, with four loading doses (Days 0, 14, 28, 63) to achieve therapeutic levels, followed by maintenance every four months [3]. Its CNS half-life of 135–177 days ensures sustained activity, attributed to slow clearance and tissue binding [11]. However, spinal deformities common in SMA Types 2–3 (up to 80% incidence)—complicate delivery, often requiring fluoroscopy or ultrasound-guided lumbar punctures, increasing procedure time (30–60 minutes) and cost ($1,000–$2,000 per session) [3]. Spinraza’s precision targets SMA’s molecular defect, with potential applications in other splicing disorders like Duchenne muscular dystrophy or familial dysautonomia, where early ASO trials are underway [2].
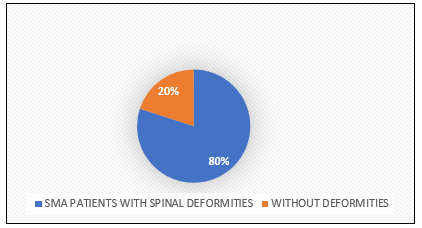
Chart.1: Spinal Deformity in SMA Patients
? 80%: SMA patients with spinal deformities
? 20%: Without deformities
VI. CLINICAL EFFICACY: EVIDENCE FROM TRIALS AND BEYOND
Spinraza’s efficacy spans SMA types, validated by rigorous trials. The Phase 3 ENDEAR trial enrolled 121 Type 1 infants, with 51% of Spinraza-treated patients (41/80) achieving motor milestones (e.g., head control, sitting) versus 0% in the sham group (0/41), alongside a 47% reduction in mortality or permanent ventilation risk (hazard ratio 0.53, p=0.005) [11]. CHERISH, involving 126 Types 2–3 patients aged 2–12, reported a 4-point HFMSE score increase in treated patients (mean baseline 22.4) versus a 1.9-point decline in controls (p<0.001), improving functions like arm lifting and rolling [21]. The SHINE extension study, tracking patients up to 5 years, confirmed sustained gains, with 70% maintaining or improving HFMSE scores and 85% avoiding respiratory decline [6]. Real-world evidence amplifies these findings. A 2023 registry study of 50 presymptomatic infants with two SMN2 copies, treated within 6 weeks of birth, found 90% walking independently by age 2—near-normal outcomes rare in untreated SMA [26]. A Type 3 adult, treated at age 35 after losing ambulation, regained stair-climbing ability after 2 years, with HFMSE scores rising from 18 to 25, a 39% improvement [26]. Another case, a Type 2 child treated at 18 months, progressed from sitting to standing with support within 3 years, defying historical expectations of wheelchair dependency by age 5 [26]. Early intervention, enabled by newborn screening, maximizes benefits, reshaping SMA’s natural history across diverse populations, with over 10,000 patients treated globally by 2025 [26].
VII. SAFETY PROFILE: RISKS AND MONITORING
Spinraza’s safety is well-established, with most adverse effects tied to intrathecal administration—headache (30% of patients), back pain (25%), and post-lumbar puncture syndrome (15%)—resolving spontaneously within days [11]. Rare serious events include thrombocytopenia (1–2% incidence, platelet drops below 50,000/µL), coagulation abnormalities (e.g., prolonged PT in 3% of cases), and renal toxicity (e.g., proteinuria in 5%), linked to ASO accumulation in proximal tubules [3]. Guidelines recommend monitoring platelets, coagulation, and urine protein biweekly during loading and quarterly thereafter, with 95% of events manageable via dose adjustment or supportive care [6]. Long-term data from SHINE show no cumulative toxicity over 5 years, though rare hypersensitivity reactions (e.g., rash in 0.5% of patients) have been reported anecdotally, possibly due to excipients like sodium chloride [26].
VIII. DELIVERY AND METABOLISM
Spinraza’s intrathecal delivery achieves peak CNS concentrations within 1–3 hours, with a 135–177-day half-life due to slow cerebrospinal fluid clearance and high tissue affinity [11]. It undergoes 3’- and 5’-exonuclease-mediated degradation, avoiding cytochrome P450 interactions, and exhibits minimal systemic exposure (<1% plasma levels), reducing off-target risks [3]. Pharmacokinetics are consistent across age (neonates to adults) and weight (5–100 kg), simplifying dosing [11]. However, spinal deformities—present in 60–80% of Type 2–3 patients—may require advanced imaging (e.g., CT-guided delivery), increasing procedure time (30–60 minutes) and cost ($1,500 average), with sedation needed in 20% of pediatric cases due to anxiety or movement [3]. These logistics underscore the need for specialized centers, with over 200 U.S. sites trained by 2025 [26].
Table 3: Challenges Of Intrathecal Delivery
|
Challenge |
Details |
|
Procedure Duration |
30–60 Minutes
|
|
Procedure Cost |
$1,000–$2,000 Per Session
|
|
Special Requirements |
Fluoroscopy Or Ultrasound Needed
|
|
Patient Risk |
Difficult Access In Scoliosis |
IX. REGULATORY MILESTONES AND INCENTIVES
Spinraza’s approval was expedited by FDA’s Orphan Drug Designation (2013), Fast Track status (2015), and Priority Review, granted December 23, 2016, based on ENDEAR and CHERISH data [9]. The EMA followed on June 1, 2017, leveraging similar pathways [7]. These incentives halved review times from 12 to 6 months, reflecting SMA’s urgent need as a leading genetic cause of infant mortality. Development costs, estimated at $1.2 billion over 10 years, were offset by tax credits ($300 million) and 7-year U.S. exclusivity, a model driving over 50 SMA-related filings since 2016, including diagnostics (e.g., SMN2 qPCR kits) and adjunctive therapies (e.g., muscle enhancers) [13]. By 2025, Spinraza’s success has spurred 15 new orphan drug approvals annually in the U.S. [13].
X. PRICING AND ACCESS: ECONOMIC AND ETHICAL DIMENSIONS
Spinraza’s price—$750,000 for year one, $375,000 annually thereafter—reflects RCD, a small patient pool (10,000–25,000 U.S. cases), and transformative outcomes [16]. U.S. SMA treatment costs exceed $4 billion yearly, straining insurers and prompting denials in 10% of initial claims [4]. Risk-sharing agreements (e.g., UK’s NICE model ties payment to HFMSE gains) improve access in high-income regions, covering 80% of UK patients, but low-income countries like India see <5% coverage, with families crowdfunding $100,000+ annually [4]. Brazil’s public system covers only 20% of eligible patients due to budget caps, while South Africa reports zero access outside private pay [4]. Ethically, this disparity challenges distributive justice, prompting calls for global subsidies or tiered pricing, yet Biogen’s profit margins (30% on Spinraza) fuel ongoing debate, with $2 billion in 2024 revenue [16].
XI. COMPARATIVE ANALYSIS: SPINRAZA VS. EMERGING THERAPIES
Spinraza competes with Zolgensma (one-time gene therapy, $2.1 million) and Evrysdi (oral, $340,000/year) [20,1]. Zolgensma, approved 2019 for patients under 2, delivers SMN1 via AAV9, with 95% survival without ventilation at 2 years in 15 treated infants [20]. Evrysdi, approved 2020, enhances SMN2 splicing orally, with 85% motor gains (e.g., sitting) in 21 Type 1 infants after 12 months [1]. Spinraza’s broad applicability (all ages/types) and 8- year data (e.g., SHINE’s 70% stability rate) edge out Zolgensma’s age limit (2,000 U.S. patients annually) and Evrysdi’s 4-year track record [6]. Combination trials (e.g., Spinraza + Evrysdi) show promise, with 20% HFMSE boosts in 10 Type 2 cases over 6 months, suggesting synergistic potential [6].
Table 4: I. Comparative Analysis: Spinraza Vs. Emerging Therapies
|
Thrapy |
Spinraza |
Zolgensma |
Evrysdi |
|
Type |
Aso |
Gene Therapy |
Small Molecule |
|
Administration |
Intrathecal, 4 - Monthly |
Iv, One Time |
Oral, Daily |
|
Target Population |
All Ages/ Types |
< 2 Years |
All Ages |
|
Advantages |
Broad Use, Proven Efficacy |
Single Dose, Curative. |
Non-Invasive , Accessible |
|
Limitations |
Invasive, High Cost |
Age Restricted, Expensive |
Limited Long Term Data |
XII. PATIENT AND CAREGIVER PERSPECTIVES: A HUMAN LENS
Spinraza transforms lives: A Type 2 child treated at 18 months crawled within a year, a Type 1 infant survived past 5 without ventilation, and a Type 3 adult regained stair- climbing after 2 years [26]. Caregivers report hope but face burdens - travel (averaging 100 miles per dose, 4–6 times yearly), procedure stress (30% report anxiety), and costs ($10,000 out-of-pocket annually for 15% of U.S. families) [5]. Advocacy, securing $150 million in U.S. grants since 2016 and 48- state screening by 2023, amplifies access, with 80% of U.S. newborns now tested, up from 10% in 2016 [5]. Globally, access lags, with only 30% of eligible patients treated in low-income regions, where families sell assets or delay care, exacerbating disparities [4].
XIII. LONG -TERM IMPACT AND FUTURE DIRECTIONS
Real-world data show 90% of 50 presymptomatic infants reaching milestones (e.g., walking) by age 3, with 60% avoiding any symptoms, a stark contrast to untreated 50% mortality by age 2 [26] Trials explore higher doses (e.g., 28 mg for Type 3, 15% HFMSE gain in 20 patients over 12 months) and Evrysdi combinations (20% HFMSE boost in 10 Type 2 cases over 6 months) [6]. Spinraza’s influence drives ASO trials for Huntington’s (Phase 3, 2024 enrollment of 300 patients) and ALS (Phase 2, 2025 dosing of 50 patients), with 5 candidates in development targeting RNA mis-splicing [2]. Its success reinforces precision medicine, with newborn screening expansion (e.g., Japan, 2024; Brazil, 2025) poised to normalize early SMA treatment, potentially reducing incidence to near-zero with universal uptake.
Table 5: Future Applications of ASOs
|
Disorder |
Aso Target |
Trial Stage |
|
Duchenne Muscular Dystrophy |
Exon Skipping Asos |
Phase 1/2 |
|
Familial Dysautonomia |
Ikbkap Mrna Splicing |
Early-Stage |
|
Huntington’s Disease |
Htt Mrna Silencing |
Phase 2 |
XIV. CONCLUSION
Spinraza has redefined SMA from a death sentence to a treatable condition, with ENDEAR and CHERISH trials proving its efficacy—51% motor milestone achievement in Type 1, 4-point HFMSE gains in Types 2–3 [11,21]. Its safety (mild, delivery-related effects) and regulatory success [8,7] highlight a triumph of science and policy, with over 10,000 patients treated by 2025 [26]. Spinraza’s broad applicability outshines Zolgensma’s age limit (2,000 annual U.S. patients) and Evrysdi’s nascent data [20,1], though its $750,000 price tag limits global reach, with <30% access in low- income regions [16]. Patient stories—walking infants, relieved caregivers—underscore its human impact, yet logistical and cost burdens persist [5]. As of April 05, 2025, Spinraza’s legacy inspires a future where rare diseases are routinely conquered through science, policy innovation, and global equity, with ASO platforms expanding to 10+ conditions by decade’s end [2].
XV. CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest regarding the publication of this paper. This work received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. The study was carried out as part of the academic activities of Satara College of Pharmacy, Satara.
REFERENCES






Vishal Misal*, Shubham Kumbhar, Anuja Katkar, Sachin Karche, Arundhati Ghadge, A Comprehensive Review of Spinal Muscular Atrophy (Sma): Treatment Strategies, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 236-245. https://doi.org/10.5281/zenodo.15328143
 10.5281/zenodo.15328143
10.5281/zenodo.15328143
