We use cookies to make sure that our website works properly, as well as some ‘optional’ cookies to personalise content and advertising, provide social media features and analyse how people use our site. Further information can be found in our Cookies policy
Lipospheres are lipid-based drug delivery systems designed to enhance the solubility and bioavailability of poorly water-soluble drugs. These colloidal carriers consist of a solid hydrophobic lipid core stabilized by a phospholipid layer, offering advantages such as high drug entrapment, controlled release, and improved drug stability. Lipospheres are particularly beneficial for the oral administration of Biopharmaceutics Classification System (BCS) Class II drugs, including cyclosporine A, by improving their dissolution rate and gastrointestinal absorption. Various preparation techniques, including high-pressure homogenization and solvent evaporation, allow for the production of lipospheres with controlled particle size and release profiles. The incorporation of biocompatible lipids and surfactants enhances their effectiveness as carriers for hydrophobic drugs, protecting them from enzymatic degradation and first-pass metabolism. Due to their stability, cost-effectiveness, and ease of scale-up, lipospheres are widely explored for pharmaceutical applications such as antibiotic, anaesthetic, vaccine, and anticancer drug delivery. Ongoing research focuses on optimizing liposphere formulations to maximize drug solubility, absorption, and therapeutic efficacy. This review highlights the role of lipospheres in drug delivery, their formulation strategies, and their potential in overcoming solubility-related challenges in pharmaceutical development.
Keywords
Lipospheres, drug delivery, solubility enhancement, controlled release, bioavailability, BCS Class II, lipid-based carriers.
Introduction
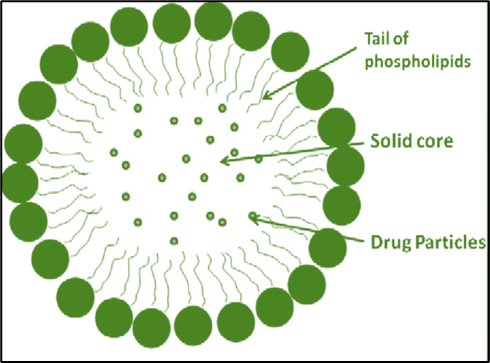
Lipospheres are lipid-based, water-dispersible solid particles ranging in diameter from 0.01 to 100 micro-meters. They are made up of a solid hydrophobic lipid core (triglycerides) that is supported by a layer of phospholipid molecules embedded in their surface. The lipospheres are appropriate by mouth, parenteral, and topical administration of drug substances and are developed to address the limitations of existing colloidal systems such as emulsions, liposomes, and polymeric nanoparticles [1,2]. Lipospheres can be more successfully spreading than most suspension-based systems, and the substances to be gave are not easily soluble in vehicle fluid because they can be dispersed in a solid medium [3,4]. The release rate of lipospheres varies in part on what is inside of the inner and exterior layers of phospholipids, which can be changed by varying the structure appropriately. The liposphere-based carrying system offers various benefits over alternative delivery systems such as emulsion liposomes and microspheres, including high stability, cheap component cost, and simple production and scalability up. High entrapment output for hydrophobic medicines, regulated particle size, and longer release of entrapped drug (5]. Lipids can be used because they are well tolerated by the body, increasing the solubility of insoluble in water drugs, allowing for larger pharmaceutical loadings, protecting the drug from impurities, and preventing biological deterioration associated with route delivery. In addition, there is regulated release. Lipospheres in the micrometer range of size appear to be more durable. After 30 days, the size of the particles and distributions remained comparable to the original formulation, while Domb and Maniar reported no evidence of aggregation in one formulation maintained at 4°C for 10 months. In-vitro release studies have shown that micro particle liposphere formulations provide longer drug release. Drug release was detected across several days, with a sudden release of pharmaceuticals occurring during the first few hours (6]. Solubility, dissolution, and intestinal permeability are basic characteristics that determine the pace and degree of drug uptake as well as bioavailability [7]. A drug's water solubility is a crucial feature that influences its uptake following taking it by mouth. It also determines the likelihood of a medication getting given parenterally and is important for modifying and evaluating drug characteristics during the drug creation and manufacturing process. Solubility of a pharmaceutical is an average metric, but the rate at which the solid drug or medication from the form of administration dissolves into solution is crucial when dissolving time is restricted 8]. While a medication's bioavailability in the mouth is determined by water solubility, drug permeability, dissolution rate, first-pass metabolism, and susceptibility to efflux mechanisms, water solubility and drug penetration are also key factors associated with oral bioavailability (9]. In drug development, the amount of insoluble pharmaceutical candidates has grown in the past few years, with about 70% of new candidates for drugs having low water solubility 10]. Weak water solubility and dissolution in GI fluids restrict these medication candidates' in vivo bioavailability following administration by mouth. As a result, laboratory-based dissolution has been acknowledged as a key aspect in drug research, and improving the rate of dissolution of poorly soluble medications while improving their bioavailability is a significant issue for pharmaceutical scientists [11, 12]. Biopharmaceutical categorization system the biopharmaceutics classification system (BCS) is a technical categorization of medicinal substances that depends on their water solubility and permeability in the gut, which connects in vitro breakdown and in vivo bioavailability of therapeutic products. [7,13].
Figure:1 Lipospheres with solid hydrophobic core surrounded by phospholipid embedded in surrounding.
When paired with the drug product's in vitro dissolving properties, BCS considers two important factors: solubility and permeability of the intestinal tract, which determine the pace and amount of drug absorption through the mouth from the solid form of dosage, as well as the product's bioavailability. Because of this, BCS is a critical technique in drug research, particularly in the creation of oral medication formulations. The FDA criterion for the solubility of a pharmaceutical categorization in BCS is based on the greatest dosage strength in an instant release (IR) orally product [14]. A drug is termed highly soluble if its greatest strength is soluble in 250 ml (this volume is taken from normal bioequivalence testing methods) or less of aqueous medium over the pH range of 1.0 to 7.5; otherwise, the drug substance is categorized as less soluble.
The permeability categorization, on the opposite hand, depends primarily on the degree of gastrointestinal uptake of a pharmacological molecule in people or indirectly on measurements of the rate of mass movement across the human gut membrane, in animals, or in vivo models [13, 14]. A pharmacological substance is deemed extremely permeable if its intestinal absorption is 90% or greater based on the balance of mass or in relation to an intravenous reference dosage. The bioavailability of BCS class II medicines is most likely restricted by their dissolving rate. However, due to their high permeability, BCS class II medicines have recently become the subject of solubility improvement research, and numerous formulating techniques for this class of molecules have been developed [15, 16, 17].
Table:1 Biopharmaceutics Classification System (BCS) with characteristics of drugs:
BCS class
Solubility
Permibility
Absorption pattern
Examples
I
High
High
Well absorbed
Metoprolol, Diltiazem, Propranolo
II
Low
High
Well absorbed
Phenytoin, Nifedipine, cyclosporine A
III
High
Low
Variable
Cimetidine, Acyclovir, Captopril
IV
Low
Low
Poorly absorbed
Hydrochlorothiazide, Taxol, Furosemide
Solubility is defined by IUPAC as the statistical composition of a solution that is saturated expressed as the amount of a specific solute in a particular solvent. Solubility can be expressed in terms of concentration, molality, mole fraction, mole ratio, and other units 18]. The widespread usage of solubility from many perspectives has resulted in solubility being stated in numerous ways. It is often stated as a concentration, either by mass (g of solute per kg of solvent, g per dL (100 mL) of solvent), molarity, molality, mole fraction, or other concentration-related terms. The greatest equilibrium quantity of solute that may dissolve per amount of solvent is the solute's solubility in that solvent given the stated criteria [19]. The partition coefficient (Log P) evaluates a compound's variable solubility in a hydrophobic solvent (octanol) and a hydrophilic solvent (water). The logarithm of both of these figures allows substances to be classified according to their hydrophilicity (or hydrophobicity). The USP and BP categorize solubility regardless of the solvent employed, but solely in terms of quantification, and have set the criteria as shown in Table 20 and 21].
Table:2 Solubility Criteria
Descriptive term
Part of solvent required per part of solute
Very soluble
Less than 1
Freely soluble
From 1 to 10
Soluble
From 10 to 30
Sparingly soluble
From 30 to 100
Slightly soluble
From 100 to 1000
Very slightly soluble
From 1000 to 10,000
Practically insoluble
10,000 and over
Importance Of Solubility:
Because of its simplicity of administration, high patient compliance, cost efficiency, minimal sterility limitations, and flexibility in dosage form design, oral ingestion is the most convenient and widely used mode of drug delivery. As a result, many generic medication companies are more likely to develop bioequivalent oral drug formulations [22]. However, the main issue in designing oral dose forms is their low bioavailability. Several variables contribute to oral bioavailability, including water solubility, drug permeability, dissolution rate, first-pass metabolism, and pre-systemic metabolism. responsiveness to efflux mechanisms. The most common reasons of insufficient oral bioavailability are poor solubility and low permeability. Solubility is also important in other dose forms, such as parenteral formulations [23]. Solubility is a crucial characteristic for attaining the necessary drug concentration in systemic circulation and eliciting the requisite pharmacological reaction (24). Insufficiently water-soluble medicines may require high dosages to achieve therapeutic plasma concentrations following their ingestion by mouth. Low water solubility is a serious issue in the formulation creation of novel chemical entities and generic drugs. Any medicine that will be absorbed must be present in the form of an aqueous solution at the absorption site. Water is the preferred solvent for liquid medicinal compositions. Most medications are either mildly acidic or weakly basic, with little water solubility. More than 40% of novel chemical entities (NCEs) generated in the pharmaceutical sector are essentially insoluble in water. These weakly water-soluble medicines have sluggish drug absorption, resulting in low and variable bioavailability and gastrointestinal mucosal toxicity. For orally given medicines, solubility is the most critical rate-limiting factor in achieving the appropriate concentration in systemic circulation for the pharmacological effect. The problem of solubility is a significant difficulty for formulation researchers 25. The increase of drug solubility and hence bioavailability through the oral route remains one of the most difficult parts of the process of drug development, particularly for mouth-drug delivery systems. There are various ways available and documented in the literature for increasing the solubility of weakly water-soluble medicines. The approaches are chosen based on specific criteria such as the qualities of the medicine under consideration, the nature of the excipients to be used, and the nature of the planned dosage form. Insufficient solubility and slow dissolution rate of weakly water-soluble medicines in watery digestive tract fluids frequently result in limited bioavailability. The bioavailability of drugs classified as class II (low solubility and high permeability) by the BCS may be increased by enhancing the drug's solubility and dissolution rate in gastrointestinal fluids. The rate-limiting step for BCS class II medications is drug release from the dosage form and solubility in the stomach fluid, not absorption; hence, increasing solubility enhances bioavailability for BCS class II pharmaceuticals 22,25,26.
Advantages Of Lipospheres:
Lipospheres offer several advantages over other means of dispersion, including [27-29].
Lack of coalescence leads to greater liposphere stability.
High aqueous dispersion.
Inexpensive ingredients.
Ease of growing up and planning.
Excessive hydrophobic drug trapping.
A drug delivery system with controlled release and particle size.
Integrated drug molecules are less mobile, reducing leakage and preventing instability induced by contacts with the emulsifier layer.
Disadvantages of lipospheres:
Furthermore, lipospheres have numerous limitations [27-29].
The presence of several lipid modifications and colloidal species can affect the melting point and solubility of active and auxiliary species.
Hydrophilic compounds have lower drug loading capacity.
Distribution dynamics is nonlinear.
High pressure might cause degradation in pharmaceuticals.
Insufficient information on stability.
Lipospheres offer multiple advantages over other particulate delivery systems such as emulsions, liposomes, and microspheres, including improved drug stability, formulation stability, the ability to freeze dry and reconstitute, controlled drug release, high drug payload, controlled particle size, the absence of carrier toxicity, and the incorporation of organic solvents. The use of lipospheres for oral delivery has several advantages, including drug protection against hydrolysis, enhanced drug bioavailability, and sustained plasma levels [30]. Furthermore, the matrix is made up of recognized physiological components and/or excipients (e.g., GRAS status), lowering the risk of acute/chronic toxicity [31]. On the opposite hand, the negative aspects of such delivery systems are primarily associated with their preparation techniques, which involve high pressure and rapid temperature changes, and include high pressure-induced drug degradation, lipid crystallization, gelation phenomena, and the coexistence of multiple colloidal species 32]. Lipospheres are now produced using a variety of processes, including high pressure homogenization, Hot and cold homogenization, solvent emulsification, evaporation, and so on [30]. An replace strategy is to create lipospheres with particles smaller than 100 nm in situ. This approach was created utilizing a dispersible pre-concentrate system 33. This delivery method, known as pro-nanoliposphere (PNL), is based on a solution including the medication, triglyceride, phospholipid, and other additives in a combination of common surfactants and an organic solvent that is soluble with all components. When this solution is carefully mixed in water-based environments, such as the upper GI lumen content, nanoparticles develop naturally. This study will concentrate on recent findings on the synthesis, physicochemical characteristics, and in vitro assessment of lipospheres and PNL as carrier systems for poorly water-soluble medicines. These lipid dispersions are suitable for a variety of administration methods. The peroral route is the most popular method of medication delivery, whereas the parenteral route is the most difficult. Thus, the focus of this study will be on nano-dispersion devices for parenteral and peroral delivery of drugs that are not water-soluble. However, administrations aimed at the eyes and the central nervous system will also be explored.
Types, Of Lipospheres:
This study summarizes the many types of lipospheres based on the particle size and matrix composition. Lipospheres are advanced drug delivery vehicles that encase medicinal chemicals in lipid-based frameworks to enable controlled ejection and bioavailability. Understanding the many types of lipospheres is vital for using them in medical compositions.
Based on matrix composition lipospheres are classified as:
This is an overview of the many types of lipospheres based on the composition of their matrix. Lipospheres are advanced delivery systems that use active compounds embedded in fat or polymer matrices.
Classical lipospheres:
These consist of a matrix made of lipids, with the majority of the lipids being neutral, which they employ to penetrate the lipophilic core, for example.
TriCaprin,
Tri Lauren,
Stearic acid,
Hydrogenated vegetable oil,
TriStearin,
Ethyl Stearate.
Polymer lipospheres:
These include matrix composed of biodegradable polymers, such as.
poly lactic acid (PLA),
polycaprolactone (PCL),
poly lacticco-glycolide (PLGA).
Polymeric matrix lipospheres are thought to be an effective tool for controlled delivery and have been studied to attain extended-release times.
Based on the size of particles:
This an overview of the types of lipospheres based on their size composition. Lipospheres are specialized delivery systems that encapsulate active ingredients within lipid size range vary nano to micrometer.
Solid lipid microparticles (SLMs):
Solid lipid particles are micro and nanoscale drug carriers made out of a matrix of solid wax heated to extremely high temperatures, fatty alcohols, and fatty acid glycerides. Solid lipid microparticles have a number of advantages for lipophilic substances as drug delivery methods; the proportion of medication included can range up to 80%, and because they are made of physiological or physiologically relevant material, they are well tolerated in biological systems. In addition to regulating medicine release and drug targeting, the solid matrix protects loaded labile chemicals from degradation [34].
General Composition of Lipospheres:
Natural degrading lipid components are used in the liposphere formulation process. Lipids, particularly triglycerides, make up the liposphere's interior hydrophobic core, while the phospholipid layer around the liposphere offers exterior function. The hydrophobic cores of the lipospheres are also created using stabilizers and neutral lipids.
Table:1 Composition of lipospheres for controlled delivery [35]
Excipients
Use
Lipid:
Stearic acid,
Glyceryl monostearate,
Glyceryl monooleate,
Ethyl stearate,
Trilaurin
Used as formation of lipid core in which drug is dispersed.
Surfactant:
Soybean phosphatidylcholine
Dimyristoyl phosphatidylglycerol
Pure egg phosphatidylcholine
The phospholipids used to form the surrounding layer of lipospheres and help in emulsification.
Stabilizers:
Pectin
Polyvinyl alcohol
Polyoxyethylene sorbitan
Used as stabilizer prevent aggregation of lipospheres.
Drugs:
Active moiety for therapeutic activity
Mechanism Of Drug Release By Orally Administered Lipid?Based Drug Delivery System:
There are mainly 4 basic principles for drug release from lipid-based formulation Solubility, Dispersion, Digestion and Absorption.
But here we are discussing 2 main principles that are:
Digestion
Absorption
Digestion:
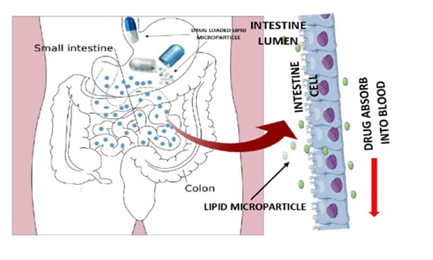
When lipid-based formulations are administered orally, gastric lipase and tongue begin to degrade the formulation's triglyceride and exogenous dietary fat. The stomach physically combines the formulation and food into the gastric content (by propulsion, grinding, and retropulsion) and generates a crude emulsion (by combining aqueous gastric fluid with lipid digestion products) similar to mate rial. Pancreatic lipase and its cofactor, co-lipase203, primarily function at the sn-1 and sn-3 positions of triglyceride to produce monoglyceride and free fatty acid, which are then broken down in the small intestine into diglyceride, monoglyceride, and fatty acids. Pancreatic phospholipase hydrolyzes phospholipids at the sn-2 position, resulting in the production of lysophosphatidic choline and fatty acid. The presence of exogenous lipids in the small intestine stimulates the release of endogenous biliary lipids from the gallbladder, which include bile salt, phospholipids and cholesterol. In the presence of bile salts, previously formed monoglycerides, fatty acids, and lyso-phospholipids (lipid digestion byproducts) are mixed to form a variety of colloidal structures, including micelles and unilamellar and multilamellar vesicles. The presence of exogenous lipids in the small intestine stimulates the production of endogenous biliary lipids from the gallbladder, such as bile salt (BS), PL, and cholesterol. In the presence of bile salts, previously formed monoglycerides, fatty acids, and Lyso-phospholipids (lipid digestion byproducts) are mixed to form a variety of colloidal forms, including micelles, unilamellar, and multilamellar vesicles. These synthesized lipid metabolites significantly improve the small intestine's capacity to solubilize and absorb lipid digestion products and medications. The following stages are involved in the digestion of lipids. 36-44] (Fig. 2)
Absorption:
Lipid-containing formulations can be used to influence medication absorption through a variety of approaches. They can stimulate lymphatic transport of medications by influencing intestinal environment [45, 46]. However, after dissolving API in the lipid-based matrix of this drug delivery system, the medication was found to have better absorption than traditional solid dosage forms [47,48]. The absorption characteristic is due to the easy wetting of hydrophobic drug particles inside the lipid matrix. This might be because the formulation contains surfactant. Similarly, the lipid matrix promotes drug entrapment in micelles [49]. Because dissolution is the rate-limiting step for the absorption of hydrophobic drugs, the primary role of these lipids is to improve the dissolution step by assembling different colloidal particles with bile components, allowing them to eventually retain a large amount of lipophilic medicament in solution via micellar solubilization 50-52. The ultimate aim of any oral lipid-based formulation is for the drug to be well absorbed by intestinal mucosal cells.
Figure:2Lipid Digestion and Drug Solubilisation Process in The Small Intestine
Figure:3 Sequential Steps of Lipid Digestion
Method Of Preparation of Lipospheres:
Lipospheres have an interior hydrophobic core made up of lipids, primarily solid triglycerides, while the surrounding phospholipid layer provides surface activity. Lipospheres have the distinct benefit of having a lipid core composed of physiologically occurring biodegradable lipids, which reduces the risk of acute and long-term toxicity. The lipid that forms the core component of the lipospheres and PNL is solid at normal temperature and may melt or remain solid at body temperature, depending on particle design.
Using solid lipid as a core may decrease or eliminate numerous drawbacks associated with the use of liquid or semi-liquid lipid cores, namely intrinsic instability and irreversible drug/excipient precipitation [53]. Typically, the oil with the highest solubilizing capability for the medication under research is chosen first, with the goal of attaining maximum drug loading in the liposphere. Currently, the selected oil should be capable of producing nano-sized particles. As a result, the selection of the oily phase is frequently a compromise between its capacity to solubilize the medication and its ability to allow the creation of a nano-encapsulation system with desirable properties. 54. Tricaprin, trilaurin, tristearin, stearic acid, ethyl stearate, and hydrogenated vegetable oil are common neutral lipids used for the hydrophobic core of liposphere compositions. Modified or hydrolyzed vegetable oils have also been frequently employed as excipients due to their superior drug solubility properties. They provide formulative and physiological benefits, and their breakdown products mirror the normal byproducts of intestinal digestion. Safety issues frequently guide the selection of an acceptable surfactant for liposphere compositions. Emulsifiers from natural sources are chosen since they are thought to be safer than synthetic surfactants. Non-ionic surfactants are less harmful than ionic surfactants, although they can cause reversible alterations in intestinal permeability. The appropriateness of the chosen surfactant for the intended route of administration, as well as its regulatory status (e.g., generally recognized as safe [GRAS] status), must be evaluated 55. The organic solvents available are determined by the route of administration, and ophthalmic and parenteral formulations have a more restricted selection. Organic solvents such as ethanol, propylene glycol (PG), and polyethylene glycol (PEG) are appropriate for oral administration because they allow for the dissolving of substantial amounts of either the hydrophilic surfactant or the hydrophobic medication in the lipid base. Alcohols and other volatile co-solvents, on the other hand, have the disadvantage of evaporating into the soft or firm gelatin capsule shells, resulting in drug precipitation. Alcohol-free formulations intended to bypass this barrier 56 have poor lipophilic drug dissolving capability 55. Triacetin is an appropriate organic sol vent because it is miscible in the oil and lipid phases and may be utilized to solubilize a hydrophobic drug 57. The surrounding layer of lipospheres is commonly made up of pure egg phosphatidylcholine, soybean phosphatidylcholine, dimyristoyl phosphatidylglycerol, and phosphatidylethanolamine.
Homogenization methods:
High shear homogenization method: High shear homogenization and ultrasound are dispersion methods that were originally utilized in the manufacture of nano-sized particle systems. However, the existence of microparticles and metal contamination issues were frequently connected with this process, prompting the development of more complex manufacturing techniques.
High pressure homogenization (HPH) method: The first step is to incorporate the medication into bulk lipid by dissolving or dispersing it in lipid melt. A high-pressure (100-2000 bar) homogenizer accelerates this liquid to a high velocity before pushing it through a tight gap. The ensuing shear stress and cavitation forces shatter the particles, reducing them to submicron size (32).
Hot homogenization method: In this approach, the active ingredient is dissolved or dispersed in a melted solid carrier, such as tristearin or polycaprolactone, and a hot buffer solution is added simultaneously with the phospholipid powder. The heated mixture is homogenized for 2-5 minutes using a homogenizer or ultrasonic probe, resulting in a homogeneous emulsion. The HPH of the resultant emulsion is then generated at a temperature higher than the core lipid's melting point. In general, greater temperatures lower particle size; nevertheless, they may also accelerate medication breakdown. To create a homogeneous dispersion of solid lipospheres, the nano-emulsion is quickly chilled to around 20?C by submerging the formulation flask in an ice/water bath while homogenization continues (31).
Cold homogenization method: This approach was created to address numerous issues with the hot homogenization method, including high temperature-induced drug degradation, the complexity of the crystallization process, which causes drug changes, and drug dispersion to the aqueous phase. The first step is to incorporate the drug into bulk lipid by dissolving or dispersing it in lipid melt, which is then rapidly cooled, resulting in homogeneous distribution of the medication throughout the lipid matrix. Next, the solid lipid matrix is ground into micron-sized particles, which are then dispersed in a cold emulsifier solution. This solution is homogenized at low temperatures to produce a nano-sized dispersion system [32]. The cold homogenization process is commonly used to produce dexamethasone (a weakly water soluble steroid) lipospheres. Dexamethasone (active component) and tristearin (lipid core) are combined in a glass flask and heated until the mixture melts. A hot phosphate buffer solution is added, along with egg phosphatidylcholine. The mixture is homogenized for a few minutes until it has a homogeneous milky consistency. To achieve a thin dispersion, the hot formulation is rapidly cooled to below 20?C and mixed continuously. Submicron lipospheres are created by extruding them through a succession of filters at a temperature 5?C higher than the melting point of the liposphere core composition. Particle size may be decreased to around 200 nm 31.
Solvent emulsification/evaporation method: Alternatively, lipospheres might be created using a solvent approach. The idea behind this process is the emulsification of a polymeric solution in an aqueous continuous phase. In this scenario, the active drug, solid carrier, and phospholipid are all dissolved in an organic solvent. The O/W emulsion is formed by agitating two immiscible liquids. This combination is further emulsified in an aqueous phase using HPH or similar homogenization method. The drug ingredient is either dissolved in the solvent system or caught in the emulsion's dispersed phase. Agitation of the system continues until the solvent divides into the aqueous phase. The organic solvent is then evaporated, and the solid is mixed with warm buffer solution until a homogeneous dispersion of lipospheres is achieved. This technique produces hardened lipospheres that contain the active moiety 31].The mean particle size is determined by the lipid content in the organic phase, and there is an inverse relationship between lipid concentration and particle size [32]. The primary issue with this procedure is the use of an organic solvent, which must be eliminated until the concentration reaches acceptable levels.
Supercritical fluid method: To prevent organic solvent contamination, the supercritical fluid approach was investigated. The lipid and drug are dissolved in a suitable organic solvent to make a solution, which is then emulsified in an aqueous phase to form an emulsion with a discontinuous phase of micelles made up of the organic solvent, drug, and the lipid. Finally, the emulsion is treated with a supercritical fluid under appropriate circumstances, resulting in the extraction of the organic solvent from the micelles and the precipitation of solid composite lipid drug nano-sized particles in the aqueous dispersion [58].
In Vitro Evaluation of Lipospheres:
Because of the colloidal size of the fragments and the complex and dynamic nature of the delivery mechanism, accurately characterizing lipospheres is a significant difficulty 58]. The following is a discussion of the most crucial parameters that require evaluation:
Particle size: It is critical to precisely predict the particle size since it may affect the in vivo efficacy of the lipid-based encapsulation system. The paragraph that follows will go into further detail regarding these effects. Photon correlation spectroscopy, also known as dynamic light scattering [59,60,61], laser diffraction [62], transmission electron microscopy [63], scanning electron microscopy, and atomic force microscopy are some of the techniques used to determine particle size. The most common methods for assessing particle size in dispersion systems are photon correlation spectroscopy and laser diffraction. The photon correlation spectroscopy technique is used to quantify particle size by analyzing the variation in scattered light intensity caused by particle motion. Because smaller particles create more intense scattering, laser diffraction determines particle size by calculating the diffraction angle based on the particle radius. Laser diffraction covers a wide range of particle sizes, from nanoscale to hundreds of micro-meters. When examining particles larger than 3 m, laser diffraction outperforms photon correlation spectroscopy since the latter cannot distinguish micron-sized particles, while being a relatively sensitive and accurate approach for defining nanoparticles. Despite the fact that the development of phase sensitive intensity difference scattering (PIDS) increased laser diffraction's capacity to detect smaller particles 61, it is suggested to use both technologies simultaneously.
Drug incorporation and loading: The loading capacity of a lipid-based drug carrier system is an important component in determining its application [64]. We must first differentiate between entrapment efficiency and loading capacity. Entrapment efficiency is defined as the proportion of drug incorporated into lipid-based particles in relation to the total quantity of drug added—that is, the percentage of drug contained in the particles vs. the percentage of drug remaining in the dispersion medium. Laser diffraction refers to the proportion of drug incorporated into lipid particles relative to the overall weight of the lipidic phase (drug + lipid). While ultracentrifugation is the simplest and fastest approach, entrapment efficiencies are not always perfect, and ultrafiltration and microdialysis are regarded to be the most reliable methods for assessment [65]. While loading capacity is the most important aspect in assessing and improving lipid-based drug carriers, entrapment efficiencies are often high, ranging from 80% to 99% depending on the drug contained. The key drivers of loading capacity are the solubility of the medicine under research in the core lipid/lipids mix, the miscibility of drug melt and lipid melt, the physical and chemical makeup of the solid lipid matrix, and the lipid's polymorphic state [66].
Crystallinity and polymorphism: A liposphere is commonly defined as a spherical liquid droplet (core) enclosed in an emulsifier shell. The resultant particles will also crystallize upon solidification and exhibit all of the properties of crystal-like substances since the major component is a solid lipid, a crystalline material. This involves the emergence of various crystalline modifications when employing polymorphic raw materials such as triglycerides, as well as a solid-liquid transition at a particular temperature (67). When polymorphic chemicals crystallize, they frequently undergo metastable changes; but when kept, they can transform into more stable forms 68.
Drug release studies from lipospheres: Drugs given to the lipospheres' solid fat core typically escape by surface erosion, biodegradation, and/or diffusion via the matrix (69). In vitro drug release studies can be utilized to assess release kinetics, especially when monitored delivery methods are under investigation. It is important to note that the release kinetics are determined by the release circumstances, such as sink/non-sink, release medium, and so on. Because of the colloidal nature of the lipospheres, release studies are complex and may be performed using a number of separation procedures, each with advantages and disadvantages (filtration, centrifugation, dialysis). Hydrophobic drugs are the greatest candidates for integration into lipospheres; nevertheless, their low intrinsic water solubility makes it difficult to maintain sink conditions, limiting the use of simple aqueous media to analyze their dissolution profiles. When paired with low analytical sensitivity and technological issues such as nonspecific drug adsorption to filters and other portions of the dissolving equipment, this might result in unreproducible and inaccurate dissolve data and release profile assessment. A simple surfactant solution or non-aqueous dissolving media can be employed to overcome this issue. However, it should be recognized that these media share just a few characteristics with the gastrointestinal environment. As a result, a revised dissolve medium that better replicates the solubilization capabilities of gastrointestinal fluids was developed to improve in vitro in vivo prediction. This medium, which often contains phospholipids and bile salts, replicates either fed or fasting gastrointestinal conditions 70].
Applications Of Lipospheres:
The use of lipospheres for topical application of N,N-diethyl-m-toluamide (DEET) insect repellent, oral administration of cyclosporin, parenteral administration of oxytetracycline and long acting local anesthetics, as well as the administration of luteinizing hormone releasing hormone (LHRH) from polymer-based lipospheres is covered in this section.
Delivery of Local Anesthetics: Because general anesthesia might have serious adverse effects, local anesthetics are preferred over general anesthesia. Even local anesthetics, which are commonly provided as an aqueous solution, are eventually absorbed into the circulation at the application site. Systemic toxicity can occur as a result of regular local anesthetic usage. A long-acting formulation that provides extended localized blocking may be useful for chronic pain relief or post-surgical pain management. Basic local anesthetics such as lidocaine and bupivacaine were delivered via lipospheres to extend their effects for several days after a single injection. As previously stated, liposphere formulations containing local anesthetics were created utilizing the melt and solvent procedures [71,72].
Delivery of Antibiotics: Many antibiotics, including ofloxacin, norfloxacin, chloramphenicol palmitate, and OTC [oxytetracycline], as well as antifungal medications like amphotericin B and nystatin, have been effectively encapsulated in lipospheres. The development of liposphere-OTC [oxytetracycline] formulations for veterinary usage [73] demonstrated the applicability of lipospheres for antibiotic administration. Parenteral OTC therapy for farm animals requires daily medicine dose over a period of days, often three to five days, in order to achieve prolonged therapeutic blood levels. There have been multiple reports of long-acting over-the-counter [oxytetracycline] formulations [74, 75, 76].
Delivery of Vaccines and Adjuvants: A number of studies have indicated that combining antigens with lipid carriers like liposomes or microparticles such as polymeric biodegradable microcapsules increase immune response [77,78]. The physical and chemical properties of the particles were connected to their ability to enhance immunogenicity under different delivery techniques. The physicochemical properties and immunogenic efficiency of numerous liposphere-vaccine combinations including a recombinant R32NS1 malaria antigen derived from Plasmodium falciparum's circumsporozoite protein as the model antigen are examined. Lipospheres were formed by carefully melting neutral fat with phospholipid and vigorously stirring the mixture in an antigen-containing water solution, resulting in the formation of a phospholipid-stabilized solid hydrophobic fat core holding the antigen when chilled (79).
Nano-lipospheres for Cell Targeting of Anticancer Drugs: The potential application of lipospheres containing paclitaxel to overcome tumor cells' resistance to the treatment was investigated 80]. It was assumed that if paclitaxel was absorbed by the cells, the encapsulation would prevent the drug from being swiftly ejected outside the cell, resulting in a fatal amount of drug content in cell plasma. Two liposphere formulations, one based on tricaprin and the other on PCL[polycaprolactone], were compared to liposomes with the same composition but no core component. The compositions were created using the solvent process and then extruded through a series of submicron filters, generating nanoparticles having A size of 200 nm. Nanolipospheres had a comparable circulation time to liposomes. To increase the duration of lipospheres in blood circulation, they were coated with polyethylene glycol with a molecular weight of 5000, which has been shown to improve blood circulation [81].
CONCLUSION:
Lipospheres have emerged as a promising drug delivery system for enhancing the solubility and bioavailability of poorly water-soluble drugs. Their unique structural composition, consisting of a solid lipid core surrounded by a phospholipid layer, offers several advantages over conventional drug delivery methods such as emulsions, liposomes, and polymeric nanoparticles. The ability of lipospheres to encapsulate hydrophobic drugs efficiently, provide controlled drug release, and improve stability makes them an attractive option for oral drug administration. A major challenge in pharmaceutical formulations is the poor solubility of many newly developed drug candidates, which limits their bioavailability and therapeutic efficacy. The Biopharmaceutical Classification System (BCS) categorizes such drugs, with Class II drugs, like cyclosporine A, being particularly challenging due to their low solubility but high permeability. Lipospheres address this issue by enhancing the dissolution rate and protecting the drug from enzymatic degradation in the gastrointestinal tract. The preparation methods for lipospheres, including high-pressure homogenization, solvent emulsification/evaporation, and supercritical fluid techniques, allow for precise control over particle size and drug loading efficiency. These techniques ensure uniform dispersion of the drug within the lipid matrix, leading to sustained drug release and improved therapeutic outcomes. Moreover, the lipid-based nature of lipospheres enhances lymphatic uptake, further improving systemic drug absorption. In vitro and in vivo studies have demonstrated the effectiveness of lipospheres in providing prolonged drug release, reducing drug degradation, and enhancing bioavailability. Their stability, ease of large-scale production, and cost-effectiveness further support their application in pharmaceutical formulations. However, certain challenges such as potential lipid polymorphism, drug leakage, and the need for optimized formulation parameters must be addressed to maximize their efficacy. Lipospheres have shown potential in various applications, including the delivery of local anesthetics, antibiotics, vaccines, and anticancer drugs. Their ability to enhance drug absorption and reduce systemic toxicity makes them a valuable platform for oral and parenteral drug delivery. Future research should focus on overcoming formulation challenges, improving large-scale production techniques, and exploring novel lipid-based carriers for enhanced drug delivery. Overall, lipospheres represent a significant advancement in drug delivery technology, offering a viable solution for enhancing the solubility and therapeutic effectiveness of poorly water-soluble drugs. Their continued development and optimization hold great promise for improving patient outcomes and expanding the range of available pharmaceutical treatments.
REFRENCES
Domb, A.J., Bergelson, L., Amselem, S., 1996. Lipospheres for controlled delivery of substances. In: Microencapsulation, Methods and Industrial Applications. Marcel Dekker, New York, p. 377
Maniar, M.H., Amselem, D., Xie, S., Burch, X., Domb, R.A.J., 1991. Characterization of lipospheres: effect of carrier and phospholipid on the loading of drug into the lipospheres. Pharm. Res., 8
Domb, Abraham J, Maniar, Manoj. Lipospheres for controlled delivery of substances. European patent EP0502119. 1996.
Rawat M, Saraf S. Lipospheres; emerging carriers for proteins and peptides. Int J Pharm Sci Nanotech. 2008; 1: 207-213.
Lee, J., Park, T. G., & Choi, H. (2000). Effect of formulation and processing variables on the characteristics of microspheres for water-soluble drugs prepared by w/o/o double emulsion solvent diffusion method. International Journal of Pharmaceutics, 196(1), 75–83. https://doi.org/10.1016/s0378-5173(99)00440-8
Domb AJ, Maniar M. Liposphere for control delivery of substances. PCT Patent Application.WO91/07171 1991.
Amidon, G. L., Lennernäs, H., Shah, V. P., & Crison, J. R. (1995). A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharmaceutical Research, 12(3), 413–420. https://doi.org/10.1023/a:1016212804288
Williams, H. D., Trevaskis, N. L., Charman, S. A., Shanker, R. M., Charman, W. N., Pouton, C. W., & Porter, C. J. H. (2013). Strategies to address low drug solubility in discovery and development. Pharmacological Reviews, 65(1), 315–499. https://doi.org/10.1124/pr.112.005660
Krishnaiah, Y. S. (2010). Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs. Journal of Bioequivalence & Bioavailability, 02(02). https://doi.org/10.4172/jbb.1000027
Kawabata, Y., Wada, K., Nakatani, M., Yamada, S., & Onoue, S. (2011). Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. International Journal of Pharmaceutics, 420(1), 1–10. https://doi.org/10.1016/j.ijpharm.2011.08.032
Hu, J., Johnston, K. P., & Williams, R. O. (2004). Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Development and Industrial Pharmacy, 30(3), 233–245. https://doi.org/10.1081/ddc-120030422
Costa, P., & Lobo, J. M. S. (2001). Modeling and comparison of dissolution profiles. European Journal of Pharmaceutical Sciences, 13(2), 123–133. https://doi.org/10.1016/s0928-0987(01)00095-1
Wagh MP, Patel JS. Biopharmaceutical classification system: scientific basis for biowaiver extensions. Int J Pharm Pharm Sci 2010;2:12e19.
Yu LX, Amidon GL, Polli JE, et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm Res 2002;19:921e925.
Kumar, S., Bhargava, D., Thakkar, A., & Arora, S. (2013). Drug Carrier Systems for Solubility Enhancement of BCS Class II Drugs: A Critical review. Critical Reviews in Therapeutic Drug Carrier Systems, 30(3), 217–256. https://doi.org/10.1615/critrevtherdrugcarriersyst.2013005964
Onoue, S., Kojo, Y., Aoki, Y., Kawabata, Y., Yamauchi, Y., & Yamada, S. (2012). Physicochemical and Pharmacokinetic Characterization of Amorphous Solid Dispersion of Tranilast with Enhanced Solubility in Gastric Fluid and Improved Oral Bioavailability. Drug Metabolism and Pharmacokinetics, 27(4), 379–387. https://doi.org/10.2133/dmpk.dmpk-11-rg-101
Urbanetz, N. A. (2006). Stabilization of solid dispersions of nimodipine and polyethylene glycol 2000. European Journal of Pharmaceutical Sciences, 28(1–2), 67–76. https://doi.org/10.1016/j.ejps.2005.12.009
M. Aulton, “Dissolution and solubility,” in Pharmaceutics: The Science of Dosage form Design, M. E. Aulton, Ed., p. 15, Churchill Livingstone, 2nd edition, 2002.
The United States Pharmacopeia, USP 30-NF 25, 2007.
British Pharmacopoeia, 2009.
Krishnaiah, Y. S. (2010c). Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs. Journal of Bioequivalence & Bioavailability, 02(02). https://doi.org/10.4172/jbb.1000027
K. H. Edward and D. Li, “Solubility,” in Drug Like Properties: Concept, Structure, Design and Methods, from ADME to Toxicity Optimization, p. 56, Elsevier, 2008.
V. R. Vemula, V. Lagishetty, and S. Lingala, “Solubility enhancement techniques,” International Journal of Pharmaceutical Sciences Review and Research, vol. 5, no. 1, pp. 41–51, 2010.
D. Sharma, M. Soni, S. Kumar, and G. D. Gupta, “Solubility enhancement—eminent role in poorly soluble drugs,” Research Journal of Pharmacy and Technology, vol. 2, no. 2, pp. 220–224, 2009.
A. Kumar, S. K. Sahoo, K. Padhee, P. S. Kochar, A. Sathapathy, and N. Pathak, “Review on solubility enhancement techniques for hydrophobic drugs,” Pharmacie Globale, vol. 3, no. 3, pp. 001–007, 2011.
Inoue, K., Aoki, Y., Hayashi, M., Kitahar, S., Tanabe, H., Kiyoki, M., Araki, H. (1995). Ex vivo anti-platelet effects of isocarbacyclin methyl ester incorporated in lipid microspheres in rabbits. Arzneim- Forsch/Drug Res; 45:980–4.
Ma, T., Wang, L., TingyuanYang, N., Wang, D., Ma, G., & Wang, S. (2014). PLGA–lipid liposphere as a promising platform for oral delivery of proteins. Colloids and Surfaces B Biointerfaces, 117, 512–519. https://doi.org/10.1016/j.colsurfb.2014.02.039
Pandit, S. S., & Patil, A. T. (2009). Formulation and in-vitro evaluation of buoyant controlled release lercanidipine lipospheres. Journal of Microencapsulation, 26(7), 635–641. Https://doi.org/10.3109/02652040802593908
Souto, E.B., Muller, R.H., 2007. Lipid nanoparticles (SLN and NLC) for drug delivery. In: Domb, A., Tabata, Y., Ravi Kumar, M.N.V., Farber, S. (Eds.), Nanoparticles for Pharmaceutical Applications. American Scientific Publishers, Stevenson Ranch, pp. 103–122.
Domb, A., 2006. Lipospheres for controlled delivery of substances. In: Benita, S. (Ed.), Microencapsulation; Methods and Industrial Applications. , 2nd ed. Taylor and Francis, Boca Raton, pp. 297–316.
Mehnert, W., & Mäder, K. (2012). Solid lipid nanoparticles. Advanced Drug Delivery Reviews, 64, 83–101. https://doi.org/10.1016/j.addr.2012.09.021
Bekerman, T., Golenser, J., & Domb, A. (2004). Cyclosporin nanoparticulate lipospheres for oral administration. Journal of Pharmaceutical Sciences, 93(5), 1264–1270. https://doi.org/10.1002/jps.20057
Singh, M. N., Hemant, K. S. Y., Ram, M., & Shivakumar, H. G. (2010). Microencapsulation: A promising technique for controlled drug delivery. Pubmed. Https://pubmed.ncbi.nlm.nih.gov/21589795
Albeer, L., Hunter, A., Lout, H., Dunlap, E., Sankpal, U., Bowman, W. P., Basha, R. & Ray, A. 2018. Combination of Mithramycin and Standard Chemotherapeutic Agents Induces Anti-proliferative activity in Ewing Sarcoma cell lines.
J.B. Cannon, M.A. Long, Emulsions, microemulsions, and lipid based drug delivery systems for drug solubilization and delivery, part II. Oral Appl. 16, 227–254 (2008)
Pouton, C. W. (2000). Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. European Journal of Pharmaceutical Sciences, 11, S93–S98. https://doi.org/10.1016/s0928-0987(00)00167-6
Hauss, D. J. (2007). Oral lipid-based formulations. Advanced Drug Delivery Reviews, 59(7), 667–676. https://doi.org/10.1016/j.addr.2007.05.006
24. C.T. Phan, Intestinal lipid absorption and transport. Front Biosci. 6(3), 299–319 (2001)
E. Ros, Intestinal absorption of triglyceride and cholesterol: Dietary and pharmacological inhibition to reduce cardiovas cular risk. Atherosclerosis 151(2), 357–379 (2000)
Kalepu, S., Manthina, M., & Padavala, V. (2013). Oral lipid-based drug delivery systems – an overview. Acta Pharmaceutica Sinica B, 3(6), 361–372. https://doi.org/10.1016/j.apsb.2013.10.001
Ghadi, R., & Dand, N. (2017). BCS class IV drugs: Highly notorious candidates for formulation development. Journal of Controlled Release, 248, 71–95. https://doi.org/10.1016/j.jconrel.2017.01.014
Wagner, D., Spahn-Langguth, H., Hanafy, A., Koggel, A., & Langguth, P. (2001). Intestinal drug efflux: formulation and food effects. Advanced Drug Delivery Reviews, 50, S13–S31. https://doi.org/10.1016/s0169-409x(01)00183-1
Gershkovich, P., & Hoffman, A. (2007). Effect of a high-fat meal on absorption and disposition of lipophilic compounds: The importance of degree of association with triglyceride-rich lipoproteins. European Journal of Pharmaceutical Sciences, 32(1), 24–32. https://doi.org/10.1016/j.ejps.2007.05.109
E.A. Fouad, M. El-Badry, G.M. Mahrous, I.A. Alsarra, Z. Alashbban, F.K. Alanazi, In vitro investigation for embedding dextromethorphan in lipids using spray drying. Dig. J. Nano mater Biostruct. 6, 1129 (2011)
Kesharwani, R., Jaiswal, P., Patel, D. K., & Yadav, P. K. (2022). Lipid-Based Drug Delivery System (LBDDS): an emerging paradigm to enhance oral bioavailability of poorly soluble drugs. Deleted Journal, 1(2), 648–663. https://doi.org/10.1007/s44174-022-00041-0
Umeyor, E. C., Kenechukwu, F. C., Ogbonna, J. D., Chime, S. A., & Attama, A. (2012). Preparation of novel solid lipid microparticles loaded with gentamicin and its evaluationin vitroandin vivo. Journal of Microencapsulation, 29(3), 296–307. https://doi.org/10.3109/02652048.2011.651495
Khan, S., Baboota, S., Ali, J., Narang, R. S., & Narang, J. K. (2015). Chlorogenic acid stabilized nanostructured lipid carriers (NLC) of atorvastatin: formulation, design and in vivo evaluation. Drug Development and Industrial Pharmacy, 42(2), 209–220. https://doi.org/10.3109/03639045.2015.1040414
H.N. Joshi, N. Shah, Review of lipids in pharmaceutical drug delivery systems: part 2. Am. Pharm. Rev. 8(5), 120 (2005)
Porter, C. J. H., Trevaskis, N. L., & Charman, W. N. (2007). Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nature Reviews Drug Discovery, 6(3), 231–248. https://doi.org/10.1038/nrd2197
Chime, A. (2013). Lipid-based drug delivery systems (LDDS): Recent advances and applications of lipids in drug delivery. African Journal of Pharmacy and Pharmacology, 7(48), 3034–3059. https://doi.org/10.5897/ajppx2013.0004
Montenegro, L., Lai, F., Offerta, A., Sarpietro, M. G., Micicchè, L., Maccioni, A. M., Valenti, D., & Fadda, A. M. (2015). From nanoemulsions to nanostructured lipid carriers: A relevant development in dermal delivery of drugs and cosmetics. Journal of Drug Delivery Science and Technology, 32, 100–112. https://doi.org/10.1016/j.jddst.2015.10.003
Tang, B., Cheng, G., Gu, J., & Xu, C. (2008). Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discovery Today, 13(13–14), 606–612. https://doi.org/10.1016/j.drudis.2008.04.006
Date, A. A., Desai, N., Dixit, R., & Nagarsenker, M. (2010). Self-Nanoemulsifying Drug Delivery Systems: Formulation Insights, applications and Advances. Nanomedicine, 5(10), 1595–1616. https://doi.org/10.2217/nnm.10.126
Gursoy, R. N., & Benita, S. (2004). Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomedicine & Pharmacotherapy, 58(3), 173–182. https://doi.org/10.1016/j.biopha.2004.02.001
Constantinides, P. P. (1995). Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharmaceutical Research, 12(11), 1561–1572. https://doi.org/10.1023/a:1016268311867
Patel, D., & Sawant, K. (2009). Self Micro-Emulsifying Drug Delivery System: formulation development and biopharmaceutical evaluation of lipophilic drugs. Current Drug Delivery, 6(4), 419–424. https://doi.org/10.2174/156720109789000519
Shukla, D., Chakraborty, S., Singh, S., & Mishra, B. (2011). Lipid-based oral multiparticulate formulations – advantages, technological advances and industrial applications. Expert Opinion on Drug Delivery, 8(2), 207–224. Https://doi.org/10.1517/17425247.2011.547469
Mukherjee, S., Ray, S., & Thakur, R. (2009b). Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian Journal of Pharmaceutical Sciences, 71(4), 349. https://doi.org/10.4103/0250-474x.57282
Bohets, N. H., Annaert, N. P., Mannens, N. G., Van Beijsterveldt, N. L., Anciaux, N. K., Verboven, N. P., Meuldermans, N. W., & Lavrijsen, N. K. (2001b). Strategies for absorption screening in drug discovery and development. Current Topics in Medicinal Chemistry, 1(5), 367–383. https://doi.org/10.2174/1568026013394886
Mehnert, W., & Mäder, K. (2012d). Solid lipid nanoparticles. Advanced Drug Delivery Reviews, 64, 83–101. https://doi.org/10.1016/j.addr.2012.09.021
Kuo, Y., & Chen, H. (2008b). Entrapment and release of saquinavir using novel cationic solid lipid nanoparticles. International Journal of Pharmaceutics. https://doi.org/10.1016/j.ijpharm.2008.08.050
Müller, R., Jacobs, C., & Kayser, O. (2001b). Nanosuspensions as particulate drug formulations in therapy. Advanced Drug Delivery Reviews, 47(1), 3–19. https://doi.org/10.1016/s0169-409x(00)00118-6
Cardiello, P. G., Samor, T., Burger, D., Hoetelmans, R., Mahanontharit, A., Ruxrungtham, K., Lange, J. M., Cooper, D. A., & Phanuphak, P. (2003b). Pharmacokinetics of Lower Doses of Saquinavir Soft-Gel Caps (800 and 1200 Mg Twice Daily) Boosted with Itraconazole in HIV-1-Positive Patients. Antiviral Therapy, 8(3), 245–249. https://doi.org/10.1177/135965350300800309
Müller, R. (2000d). Solid lipid nanoparticles (SLN) for controlled drug delivery â?“ a review of the state of the art. European Journal of Pharmaceutics and Biopharmaceutics, 50(1), 161–177. https://doi.org/10.1016/s0939-6411(00)00087-4
Li, H., Zhao, X., Ma, Y., Zhai, G., Li, L., & Lou, H. (2008). Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. Journal of Controlled Release, 133(3), 238–244. https://doi.org/10.1016/j.jconrel.2008.10.002
Muchow, M., Maincent, P., & Müller, R. H. (2008). Lipid Nanoparticles with a Solid Matrix (SLN®, NLC®, LDC®) for Oral Drug Delivery. Drug Development and Industrial Pharmacy, 34(12), 1394–1405. https://doi.org/10.1080/03639040802130061
Bunjes, H. (2010). Lipid nanoparticles for the delivery of poorly water-soluble drugs. Journal of Pharmacy and Pharmacology, 62(11), 1637–1645. https://doi.org/10.1111/j.2042-7158.2010.01024.x
Freitas, C., & Müller, R. (1999). Correlation between long-term stability of solid lipid nanoparticles (SLNTM) and crystallinity of the lipid phase. European Journal of Pharmaceutics and Biopharmaceutics, 47(2), 125–132. https://doi.org/10.1016/s0939-6411(98)00074-5
Sawant, K., & Dodiya, S. (2008). Recent advances and patents on solid lipid nanoparticles. Recent Patents on Drug Delivery & Formulation, 2(2), 120–135. https://doi.org/10.2174/187221108784534081
Hunskaar, S., Fasmer, O. B., & Hole, K. (1985). Formalin test in mice, a useful technique for evaluating mild analgesics. Journal of Neuroscience Methods, 14(1), 69–76. https://doi.org/10.1016/0165-0270(85)90116-5
Domb, A. J. (1995). Long acting injectable oxytetracycline-liposphere formulations. International Journal of Pharmaceutics, 124(2), 271–278. https://doi.org/10.1016/0378-5173(95)00098-4
Oukessou M, Uccelli-Thomas V, Toutain PL. Pharmacokinetics and local tolerance of a long-acting oxytetracycline formulation in camels. Am J Vet Res 1992; 53: 1658–1662.
Landoni MF, Errecalde JO. Tissue concentrations of a long-acting oxytetracycline formulation after intramuscular administration in cattle. Rev Sci Tech 1992; 11:909–915.
Adawa, D., Hassan, A., Abdullah, S., Ogunkoya, A., Adeyanju, J., & Okoro, J. (1992). Clinical trial of long?acting oxytetracycline and piroxicam in the treatment of canine ehrlichosis. Veterinary Quarterly, 14(3), 118–120. https://doi.org/10.1080/01652176.1992.9694345
Alving, C. R. (1989). Liposomes as carriers of vaccines. In Progress in vaccinology (pp. 429–437). https://doi.org/10.1007/978-1-4612-3508-8_41
Eldridge, J. H., Staas, J. K., Meulbroek, J. A., McGhee, J. R., Tice, T. R., & Gilley, R. M. (1991). Biodegradable microspheres as a vaccine delivery system. Molecular Immunology, 28(3), 287–294. https://doi.org/10.1016/0161-5890(91)90076-v
Amselem S, Alving C, Domb A. Lipospheres for the delivery of vaccines. In: Bernstein H, Cohen S, eds. Microparticulate Systems for Drug Delivery. New York, NY: Marcel Dekker Inc., 1993:399–434.
Gur A. Taxol incorporated in nanoliposphere formulations against taxol resistant cells. M.Sc. thesis, The Hebrew University of Jerusalem, Jerusalem, Israel, 1994.
Lichtman-Teomim L. Injectable systems for the delivery of insoluble anticancer agents. M.Sc. thesis, The Hebrew University of Jerusalem, Israel, 1994.
Woodle, M., Newman, & Martin, F. (1992). Liposome leakage and blood circulation: Comparison of adsorbed block copolymers with covalent attachment of PEG. International Journal of Pharmaceutics, 88(1–3), 327–334. https://doi.org/10.1016/0378-5173(92)90331-u.
Reference
Domb, A.J., Bergelson, L., Amselem, S., 1996. Lipospheres for controlled delivery of substances. In: Microencapsulation, Methods and Industrial Applications. Marcel Dekker, New York, p. 377
Maniar, M.H., Amselem, D., Xie, S., Burch, X., Domb, R.A.J., 1991. Characterization of lipospheres: effect of carrier and phospholipid on the loading of drug into the lipospheres. Pharm. Res., 8
Domb, Abraham J, Maniar, Manoj. Lipospheres for controlled delivery of substances. European patent EP0502119. 1996.
Rawat M, Saraf S. Lipospheres; emerging carriers for proteins and peptides. Int J Pharm Sci Nanotech. 2008; 1: 207-213.
Lee, J., Park, T. G., & Choi, H. (2000). Effect of formulation and processing variables on the characteristics of microspheres for water-soluble drugs prepared by w/o/o double emulsion solvent diffusion method. International Journal of Pharmaceutics, 196(1), 75–83. https://doi.org/10.1016/s0378-5173(99)00440-8
Domb AJ, Maniar M. Liposphere for control delivery of substances. PCT Patent Application.WO91/07171 1991.
Amidon, G. L., Lennernäs, H., Shah, V. P., & Crison, J. R. (1995). A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharmaceutical Research, 12(3), 413–420. https://doi.org/10.1023/a:1016212804288
Williams, H. D., Trevaskis, N. L., Charman, S. A., Shanker, R. M., Charman, W. N., Pouton, C. W., & Porter, C. J. H. (2013). Strategies to address low drug solubility in discovery and development. Pharmacological Reviews, 65(1), 315–499. https://doi.org/10.1124/pr.112.005660
Krishnaiah, Y. S. (2010). Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs. Journal of Bioequivalence & Bioavailability, 02(02). https://doi.org/10.4172/jbb.1000027
Kawabata, Y., Wada, K., Nakatani, M., Yamada, S., & Onoue, S. (2011). Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. International Journal of Pharmaceutics, 420(1), 1–10. https://doi.org/10.1016/j.ijpharm.2011.08.032
Hu, J., Johnston, K. P., & Williams, R. O. (2004). Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Development and Industrial Pharmacy, 30(3), 233–245. https://doi.org/10.1081/ddc-120030422
Costa, P., & Lobo, J. M. S. (2001). Modeling and comparison of dissolution profiles. European Journal of Pharmaceutical Sciences, 13(2), 123–133. https://doi.org/10.1016/s0928-0987(01)00095-1
Wagh MP, Patel JS. Biopharmaceutical classification system: scientific basis for biowaiver extensions. Int J Pharm Pharm Sci 2010;2:12e19.
Yu LX, Amidon GL, Polli JE, et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm Res 2002;19:921e925.
Kumar, S., Bhargava, D., Thakkar, A., & Arora, S. (2013). Drug Carrier Systems for Solubility Enhancement of BCS Class II Drugs: A Critical review. Critical Reviews in Therapeutic Drug Carrier Systems, 30(3), 217–256. https://doi.org/10.1615/critrevtherdrugcarriersyst.2013005964
Onoue, S., Kojo, Y., Aoki, Y., Kawabata, Y., Yamauchi, Y., & Yamada, S. (2012). Physicochemical and Pharmacokinetic Characterization of Amorphous Solid Dispersion of Tranilast with Enhanced Solubility in Gastric Fluid and Improved Oral Bioavailability. Drug Metabolism and Pharmacokinetics, 27(4), 379–387. https://doi.org/10.2133/dmpk.dmpk-11-rg-101
Urbanetz, N. A. (2006). Stabilization of solid dispersions of nimodipine and polyethylene glycol 2000. European Journal of Pharmaceutical Sciences, 28(1–2), 67–76. https://doi.org/10.1016/j.ejps.2005.12.009
M. Aulton, “Dissolution and solubility,” in Pharmaceutics: The Science of Dosage form Design, M. E. Aulton, Ed., p. 15, Churchill Livingstone, 2nd edition, 2002.
The United States Pharmacopeia, USP 30-NF 25, 2007.
British Pharmacopoeia, 2009.
Krishnaiah, Y. S. (2010c). Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs. Journal of Bioequivalence & Bioavailability, 02(02). https://doi.org/10.4172/jbb.1000027
K. H. Edward and D. Li, “Solubility,” in Drug Like Properties: Concept, Structure, Design and Methods, from ADME to Toxicity Optimization, p. 56, Elsevier, 2008.
V. R. Vemula, V. Lagishetty, and S. Lingala, “Solubility enhancement techniques,” International Journal of Pharmaceutical Sciences Review and Research, vol. 5, no. 1, pp. 41–51, 2010.
D. Sharma, M. Soni, S. Kumar, and G. D. Gupta, “Solubility enhancement—eminent role in poorly soluble drugs,” Research Journal of Pharmacy and Technology, vol. 2, no. 2, pp. 220–224, 2009.
A. Kumar, S. K. Sahoo, K. Padhee, P. S. Kochar, A. Sathapathy, and N. Pathak, “Review on solubility enhancement techniques for hydrophobic drugs,” Pharmacie Globale, vol. 3, no. 3, pp. 001–007, 2011.
Inoue, K., Aoki, Y., Hayashi, M., Kitahar, S., Tanabe, H., Kiyoki, M., Araki, H. (1995). Ex vivo anti-platelet effects of isocarbacyclin methyl ester incorporated in lipid microspheres in rabbits. Arzneim- Forsch/Drug Res; 45:980–4.
Ma, T., Wang, L., TingyuanYang, N., Wang, D., Ma, G., & Wang, S. (2014). PLGA–lipid liposphere as a promising platform for oral delivery of proteins. Colloids and Surfaces B Biointerfaces, 117, 512–519. https://doi.org/10.1016/j.colsurfb.2014.02.039
Pandit, S. S., & Patil, A. T. (2009). Formulation and in-vitro evaluation of buoyant controlled release lercanidipine lipospheres. Journal of Microencapsulation, 26(7), 635–641. Https://doi.org/10.3109/02652040802593908
Souto, E.B., Muller, R.H., 2007. Lipid nanoparticles (SLN and NLC) for drug delivery. In: Domb, A., Tabata, Y., Ravi Kumar, M.N.V., Farber, S. (Eds.), Nanoparticles for Pharmaceutical Applications. American Scientific Publishers, Stevenson Ranch, pp. 103–122.
Domb, A., 2006. Lipospheres for controlled delivery of substances. In: Benita, S. (Ed.), Microencapsulation; Methods and Industrial Applications. , 2nd ed. Taylor and Francis, Boca Raton, pp. 297–316.
Mehnert, W., & Mäder, K. (2012). Solid lipid nanoparticles. Advanced Drug Delivery Reviews, 64, 83–101. https://doi.org/10.1016/j.addr.2012.09.021
Bekerman, T., Golenser, J., & Domb, A. (2004). Cyclosporin nanoparticulate lipospheres for oral administration. Journal of Pharmaceutical Sciences, 93(5), 1264–1270. https://doi.org/10.1002/jps.20057
Singh, M. N., Hemant, K. S. Y., Ram, M., & Shivakumar, H. G. (2010). Microencapsulation: A promising technique for controlled drug delivery. Pubmed. Https://pubmed.ncbi.nlm.nih.gov/21589795
Albeer, L., Hunter, A., Lout, H., Dunlap, E., Sankpal, U., Bowman, W. P., Basha, R. & Ray, A. 2018. Combination of Mithramycin and Standard Chemotherapeutic Agents Induces Anti-proliferative activity in Ewing Sarcoma cell lines.
J.B. Cannon, M.A. Long, Emulsions, microemulsions, and lipid based drug delivery systems for drug solubilization and delivery, part II. Oral Appl. 16, 227–254 (2008)
Pouton, C. W. (2000). Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. European Journal of Pharmaceutical Sciences, 11, S93–S98. https://doi.org/10.1016/s0928-0987(00)00167-6
Hauss, D. J. (2007). Oral lipid-based formulations. Advanced Drug Delivery Reviews, 59(7), 667–676. https://doi.org/10.1016/j.addr.2007.05.006
24. C.T. Phan, Intestinal lipid absorption and transport. Front Biosci. 6(3), 299–319 (2001)
E. Ros, Intestinal absorption of triglyceride and cholesterol: Dietary and pharmacological inhibition to reduce cardiovas cular risk. Atherosclerosis 151(2), 357–379 (2000)
Kalepu, S., Manthina, M., & Padavala, V. (2013). Oral lipid-based drug delivery systems – an overview. Acta Pharmaceutica Sinica B, 3(6), 361–372. https://doi.org/10.1016/j.apsb.2013.10.001
Ghadi, R., & Dand, N. (2017). BCS class IV drugs: Highly notorious candidates for formulation development. Journal of Controlled Release, 248, 71–95. https://doi.org/10.1016/j.jconrel.2017.01.014
Wagner, D., Spahn-Langguth, H., Hanafy, A., Koggel, A., & Langguth, P. (2001). Intestinal drug efflux: formulation and food effects. Advanced Drug Delivery Reviews, 50, S13–S31. https://doi.org/10.1016/s0169-409x(01)00183-1
Gershkovich, P., & Hoffman, A. (2007). Effect of a high-fat meal on absorption and disposition of lipophilic compounds: The importance of degree of association with triglyceride-rich lipoproteins. European Journal of Pharmaceutical Sciences, 32(1), 24–32. https://doi.org/10.1016/j.ejps.2007.05.109
E.A. Fouad, M. El-Badry, G.M. Mahrous, I.A. Alsarra, Z. Alashbban, F.K. Alanazi, In vitro investigation for embedding dextromethorphan in lipids using spray drying. Dig. J. Nano mater Biostruct. 6, 1129 (2011)
Kesharwani, R., Jaiswal, P., Patel, D. K., & Yadav, P. K. (2022). Lipid-Based Drug Delivery System (LBDDS): an emerging paradigm to enhance oral bioavailability of poorly soluble drugs. Deleted Journal, 1(2), 648–663. https://doi.org/10.1007/s44174-022-00041-0
Umeyor, E. C., Kenechukwu, F. C., Ogbonna, J. D., Chime, S. A., & Attama, A. (2012). Preparation of novel solid lipid microparticles loaded with gentamicin and its evaluationin vitroandin vivo. Journal of Microencapsulation, 29(3), 296–307. https://doi.org/10.3109/02652048.2011.651495
Khan, S., Baboota, S., Ali, J., Narang, R. S., & Narang, J. K. (2015). Chlorogenic acid stabilized nanostructured lipid carriers (NLC) of atorvastatin: formulation, design and in vivo evaluation. Drug Development and Industrial Pharmacy, 42(2), 209–220. https://doi.org/10.3109/03639045.2015.1040414
H.N. Joshi, N. Shah, Review of lipids in pharmaceutical drug delivery systems: part 2. Am. Pharm. Rev. 8(5), 120 (2005)
Porter, C. J. H., Trevaskis, N. L., & Charman, W. N. (2007). Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nature Reviews Drug Discovery, 6(3), 231–248. https://doi.org/10.1038/nrd2197
Chime, A. (2013). Lipid-based drug delivery systems (LDDS): Recent advances and applications of lipids in drug delivery. African Journal of Pharmacy and Pharmacology, 7(48), 3034–3059. https://doi.org/10.5897/ajppx2013.0004
Montenegro, L., Lai, F., Offerta, A., Sarpietro, M. G., Micicchè, L., Maccioni, A. M., Valenti, D., & Fadda, A. M. (2015). From nanoemulsions to nanostructured lipid carriers: A relevant development in dermal delivery of drugs and cosmetics. Journal of Drug Delivery Science and Technology, 32, 100–112. https://doi.org/10.1016/j.jddst.2015.10.003
Tang, B., Cheng, G., Gu, J., & Xu, C. (2008). Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discovery Today, 13(13–14), 606–612. https://doi.org/10.1016/j.drudis.2008.04.006
Date, A. A., Desai, N., Dixit, R., & Nagarsenker, M. (2010). Self-Nanoemulsifying Drug Delivery Systems: Formulation Insights, applications and Advances. Nanomedicine, 5(10), 1595–1616. https://doi.org/10.2217/nnm.10.126
Gursoy, R. N., & Benita, S. (2004). Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomedicine & Pharmacotherapy, 58(3), 173–182. https://doi.org/10.1016/j.biopha.2004.02.001
Constantinides, P. P. (1995). Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharmaceutical Research, 12(11), 1561–1572. https://doi.org/10.1023/a:1016268311867
Patel, D., & Sawant, K. (2009). Self Micro-Emulsifying Drug Delivery System: formulation development and biopharmaceutical evaluation of lipophilic drugs. Current Drug Delivery, 6(4), 419–424. https://doi.org/10.2174/156720109789000519
Shukla, D., Chakraborty, S., Singh, S., & Mishra, B. (2011). Lipid-based oral multiparticulate formulations – advantages, technological advances and industrial applications. Expert Opinion on Drug Delivery, 8(2), 207–224. Https://doi.org/10.1517/17425247.2011.547469
Mukherjee, S., Ray, S., & Thakur, R. (2009b). Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian Journal of Pharmaceutical Sciences, 71(4), 349. https://doi.org/10.4103/0250-474x.57282
Bohets, N. H., Annaert, N. P., Mannens, N. G., Van Beijsterveldt, N. L., Anciaux, N. K., Verboven, N. P., Meuldermans, N. W., & Lavrijsen, N. K. (2001b). Strategies for absorption screening in drug discovery and development. Current Topics in Medicinal Chemistry, 1(5), 367–383. https://doi.org/10.2174/1568026013394886
Mehnert, W., & Mäder, K. (2012d). Solid lipid nanoparticles. Advanced Drug Delivery Reviews, 64, 83–101. https://doi.org/10.1016/j.addr.2012.09.021
Kuo, Y., & Chen, H. (2008b). Entrapment and release of saquinavir using novel cationic solid lipid nanoparticles. International Journal of Pharmaceutics. https://doi.org/10.1016/j.ijpharm.2008.08.050
Müller, R., Jacobs, C., & Kayser, O. (2001b). Nanosuspensions as particulate drug formulations in therapy. Advanced Drug Delivery Reviews, 47(1), 3–19. https://doi.org/10.1016/s0169-409x(00)00118-6
Cardiello, P. G., Samor, T., Burger, D., Hoetelmans, R., Mahanontharit, A., Ruxrungtham, K., Lange, J. M., Cooper, D. A., & Phanuphak, P. (2003b). Pharmacokinetics of Lower Doses of Saquinavir Soft-Gel Caps (800 and 1200 Mg Twice Daily) Boosted with Itraconazole in HIV-1-Positive Patients. Antiviral Therapy, 8(3), 245–249. https://doi.org/10.1177/135965350300800309
Müller, R. (2000d). Solid lipid nanoparticles (SLN) for controlled drug delivery â?“ a review of the state of the art. European Journal of Pharmaceutics and Biopharmaceutics, 50(1), 161–177. https://doi.org/10.1016/s0939-6411(00)00087-4
Li, H., Zhao, X., Ma, Y., Zhai, G., Li, L., & Lou, H. (2008). Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. Journal of Controlled Release, 133(3), 238–244. https://doi.org/10.1016/j.jconrel.2008.10.002
Muchow, M., Maincent, P., & Müller, R. H. (2008). Lipid Nanoparticles with a Solid Matrix (SLN®, NLC®, LDC®) for Oral Drug Delivery. Drug Development and Industrial Pharmacy, 34(12), 1394–1405. https://doi.org/10.1080/03639040802130061
Bunjes, H. (2010). Lipid nanoparticles for the delivery of poorly water-soluble drugs. Journal of Pharmacy and Pharmacology, 62(11), 1637–1645. https://doi.org/10.1111/j.2042-7158.2010.01024.x
Freitas, C., & Müller, R. (1999). Correlation between long-term stability of solid lipid nanoparticles (SLNTM) and crystallinity of the lipid phase. European Journal of Pharmaceutics and Biopharmaceutics, 47(2), 125–132. https://doi.org/10.1016/s0939-6411(98)00074-5
Sawant, K., & Dodiya, S. (2008). Recent advances and patents on solid lipid nanoparticles. Recent Patents on Drug Delivery & Formulation, 2(2), 120–135. https://doi.org/10.2174/187221108784534081
Hunskaar, S., Fasmer, O. B., & Hole, K. (1985). Formalin test in mice, a useful technique for evaluating mild analgesics. Journal of Neuroscience Methods, 14(1), 69–76. https://doi.org/10.1016/0165-0270(85)90116-5
Domb, A. J. (1995). Long acting injectable oxytetracycline-liposphere formulations. International Journal of Pharmaceutics, 124(2), 271–278. https://doi.org/10.1016/0378-5173(95)00098-4
Oukessou M, Uccelli-Thomas V, Toutain PL. Pharmacokinetics and local tolerance of a long-acting oxytetracycline formulation in camels. Am J Vet Res 1992; 53: 1658–1662.
Landoni MF, Errecalde JO. Tissue concentrations of a long-acting oxytetracycline formulation after intramuscular administration in cattle. Rev Sci Tech 1992; 11:909–915.
Adawa, D., Hassan, A., Abdullah, S., Ogunkoya, A., Adeyanju, J., & Okoro, J. (1992). Clinical trial of long?acting oxytetracycline and piroxicam in the treatment of canine ehrlichosis. Veterinary Quarterly, 14(3), 118–120. https://doi.org/10.1080/01652176.1992.9694345
Alving, C. R. (1989). Liposomes as carriers of vaccines. In Progress in vaccinology (pp. 429–437). https://doi.org/10.1007/978-1-4612-3508-8_41
Eldridge, J. H., Staas, J. K., Meulbroek, J. A., McGhee, J. R., Tice, T. R., & Gilley, R. M. (1991). Biodegradable microspheres as a vaccine delivery system. Molecular Immunology, 28(3), 287–294. https://doi.org/10.1016/0161-5890(91)90076-v
Amselem S, Alving C, Domb A. Lipospheres for the delivery of vaccines. In: Bernstein H, Cohen S, eds. Microparticulate Systems for Drug Delivery. New York, NY: Marcel Dekker Inc., 1993:399–434.
Gur A. Taxol incorporated in nanoliposphere formulations against taxol resistant cells. M.Sc. thesis, The Hebrew University of Jerusalem, Jerusalem, Israel, 1994.
Lichtman-Teomim L. Injectable systems for the delivery of insoluble anticancer agents. M.Sc. thesis, The Hebrew University of Jerusalem, Israel, 1994.
Woodle, M., Newman, & Martin, F. (1992). Liposome leakage and blood circulation: Comparison of adsorbed block copolymers with covalent attachment of PEG. International Journal of Pharmaceutics, 88(1–3), 327–334. https://doi.org/10.1016/0378-5173(92)90331-u.
Neeraj Kumar Rathour
Corresponding author
Department of Pharmacy, I.E.T., M.J.P. Rohilkhand University, Bareilly
Neeraj Kumar Rathour*, Ajay Pratap Singh, Saurabh Mishra, A Review on Orally Administration of Lipospheres for Enhancement of Solubility, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 3, 1634-1652. https://doi.org/10.5281/zenodo.15044972


 10.5281/zenodo.15044972
10.5281/zenodo.15044972
