Pulla Reddy Institute of Pharmacy, Department of Pharmacy practice, Dundigal Hyderabad.
Fibrodysplasia ossificans progressiva (FOP) also known as stone man syndrome is a rare genetic disorder characterized by abnormal bone development. It is caused by activating mutations of the ACVR-1 gene. FOP is an incredibly rare disease with only around 800 cases confirmed world -wide as of 2017 and due to its rarity, awareness and understanding are lacking, leading to delayed diagnosis. This disorder is primarily described in young children and equally affects both sexes. Ossifications typically begin to develop within the first ten years of life, causing significant loss of mobility and worsening of the disease. There is no effective treatment for FOP; Potential treatment may be based on future inventions that block ACVR1gene. Early diagnosis and timely management of symptoms can significantly improve a patient's quality of life. Doctors can only manage the symptoms of the condition by helping patients maintain mobility through physical therapy, pain relief, and, when necessary, surgery. This article includes Genetics, epidemiology, pathophysiology, diagnosis and treatment of disease. (FOP).
The ?brodysplasia is a rare autosomal dominant genetic condition which is commonly known as ossi?cans progressiva (or) stoneman syndrome is characterized by the abnormal formation of bone in parts of the body where bone is not ordinarily present. [1]. It was initially named in 1970 by victor A. Most often detected in early childhood, it impacts nearly all organs [2]. The illness, which affects around one in two million people, can become very disability. The people with FOP face significant challenges with a median life expectancy of around 40 years [1].A gene mutation was the cause of FOP in the majority of the cases, but in some cases, the mutation had been passed down from an affected parent [2].under severe conditions, it may result in joints that are permanently frozen in place, making the body stone like inability to move, hence the term "stone man syndrome" [2]. In stoneman syndrome, abnormal bone growth happens on its own, but it frequently starts with viral disease-induced soft tissues injury. Tendons, ligaments, skeletal muscle tissue, connective tissue like fascia and muscle ?bres are frequently shown to exhibit abnormal bone development and these areas may have stiffness and pain [3].It is caused by activating mutations in the receptor for bone morphogenetics proteins, Activin receptor A type l (ACVR1) .FOP is characterized by genetic bilateral hallux valgus abnormalities, which impact both the postnatal and developed phases [4].Excessive bone growth may result in decreased mobility in the joints , as well as it causes di?culty in speaking and eating because of restricted mouth opening. Bone development around the rib cage may cause breathing problems by limiting lung expansion [5]. In FOP, activating mutations in ACVR1, the gene encoding the ALK2 BMP type l receptor, lead to the development of extraskeletal bone by endochondral ossi?cation and increase the activation of the bone morphogenetic protein (BMP) signaling pathway [6]. The Ossi?cations usually present develop during the ?rst ten years of life, leading to signi?cant loss and increasing disesase. There is currently no proven cure or preventative therapy [7]. Because of its rarity, this condition is not well known, which can result in an improper or delayed diagnosis [1].
Historical Background:

Fig:1
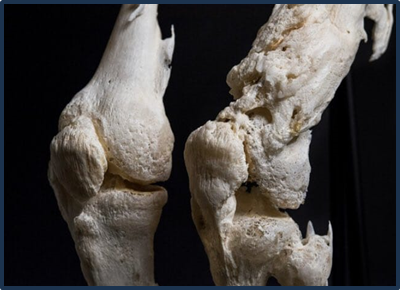
Fig:2
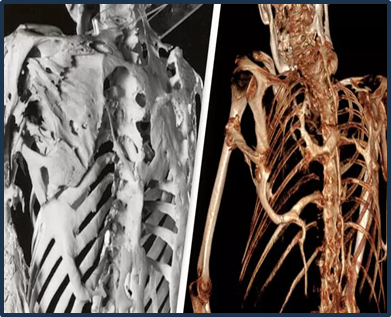
Fig:3
Genetics:
A mutation in the chromosome 2 gene ACVR1/ALK2, which is involved in the bone morphogenetic protein (BMP) signaling system, is the main cause of FOP, a morphogenetic disorder. This gene generates the transmembrane serine/ threonine kinase receptor, which interacts with bone matrix- based BMPs. In skeletal muscles, the presence of BMPs intiates the development of heterotopic bone. The majority of FOP cases are caused by a nucleotide mutation in the ACVR1/AVK2 gene at codon position 617, where adenine is substituted by guanine. Histidine takes the place of arginine at codon position 206 in the ALK2 protein as a result of this mutation [4]
Epidemiology:
FOP is an uncommon genetic condition marked by increasing bone growth in skeletal muscles and congenital abnormalities in the great toes, thumbs and vertebrae [4] as of 2017, just about 800 cases of FOP had been con?rmed globally, making it an extremely rare disease. FOP is regarded as one of the rarest diseases in human history. FOP had an estimated frequency of 0.5 cases per million people, yet it affects persons of all ethnicities despite its low prevalence [5]. It’s crucial to remember that these ?gures are still accurate and applicable today. FOP has no racial, ethnic, gender, or geographical predispositions and affects people of all ethnic backgrounds. It is an autosomal dominant trait that is transmitted with variable expression and frequently appears sporadically. FOP typically starts in early childhood and gets worse as time goes on. Because it’s so hard to treat, the Japanese Ministry of Health, Labour, and Welfare recognized it in 2007 as one of the most difficult conditions to manage. People with Fibrodysplasia Ossificans Progressiva (FOP) usually live to around 40 years of age. [10]
Pathophysiology:
The pathophysiology of FOP has become more better understood in recent years, especially since the causing missense mutations (R206H) in the ACVR1 (activin receptor type 1) gene were identified. Around 97% of all cases of FOP are caused by this autosomal dominant mutation, which have a de novo. They despite having the same mutation in patients' beginning ages and rates of illness progression differ greatly, for reasons that are not fully understood. All of the additional mutations that have been reported impact the ACVR1 gene, and their variant/atypical phenotypes range from extremely mild to more severe, including atypical features like cataracts and cerebellar abnormalities. Activin receptor type 1/activin receptor-like kinase 2 (ACVR1/ALK2) gains function as a result of the mutation, becoming hypersensitive to activin A. When ACVR1/ALK2 activated, SMAD 1/5/9 is phosphorylated to which causes the activation of transcription factors leading to heterotopic bone formation. A poorly known mechanism of inflammation and hypoxia that involves HIF, mast cells, and different inflammatory factors causes this process.[11]
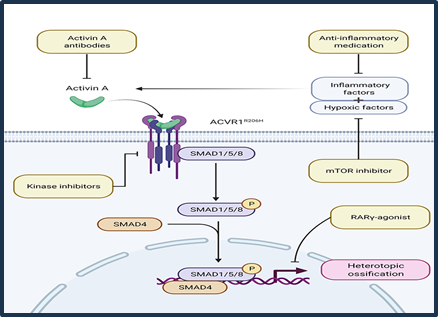
Fig;4
Symptoms:
Complications:
Treatment:
Currently, there is no cure for genetic disorders like fibrodysplasia ossificans progressiva (FOP). Surgery to remove the abnormal bone is not advised, as it can trigger the growth of new, more painful bone in the affected area. To manage this condition, doctors have developed medications that help slow down the progression and severity of bone formation. These treatments focus on reducing inflammation and flare-ups while preventing further complications.[5]
Supportive medical management was provided, including counseling the patient to rest and avoid triggering events or strenuous physical activity. [7]. The ?rst line of treatment for symptoms will involve corticosteroids, started within the initial 24-hour postpartum period, may help decrease the effects of tissue edema and in?ammation that are frequently observed in the early stages of the illness [10].
Other Names:
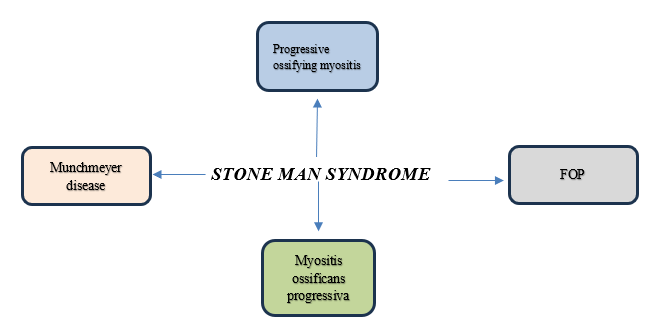
Diagnosis:
The possible conditions to consider include progressive osseous heteroplasia, osteosarcoma, lymphedema, soft tissue sarcoma, desmoid tumors (also called aggressive fibromatosis), aggressive juvenile fibromatosis, and acquired heterotopic ossification.[5]. FOP, or Stone Man Syndrome, is often misdiagnosed because its symptoms are similar to those of other genetic disorders. The most accurate way to confirm FOP is through genetic testing to check for mutations in the ACVR1 gene.[3] FOP is a rare and disabling disease with no cure or treatment to stop its progression. Early signs, such as malformed great toes, can help identify it and prevent complications later. Diagnosis relies mainly on imaging tests: Bone scans detect abnormal bone growth (heterotopic ossification). MRI scans can identify the condition early, before bone formation starts. Early diagnosis helps avoid invasive procedures, like biopsies or injections, which can worsen the condition by causing inflammation. Radiologists must understand the disease to avoid these harmful tests. Physical exams may reveal tenderness in visible lumps and stiffness in abdominal and spinal muscles.[10]
DISCUSSION:
According to mendelian inheritance in mutation (MIM)#135100, FOP is fibro dysplasia ossificans progressive. This heritable condition of connective tissue causes severe disability and is defined by congential deformities of the great toes, such as hallux valgus, deformed first metatarsal, and/or monophalangism [12]. Clinical diagnosis of FOP is typically based on the presence of three main criteria (6,14): progressive heterotopic endochondral ossification, congential great toe deformity, and temporal patterns. The erythrocyte sedimentation rate may subtly increase during flare-ups, according to laboratory examinations. A conforming test for mutations in the ACVR1 gene is genetic analysis. The diagnosis can be confirmed using imaging tests like CT scan and radiography [13], which display the million is affected by the exceeding rare genetic diseases known as MOP. First described by PATIN in 1648. 41%of MOP is screened before the age of two, 80?fore the age of ten, and 95?fore the age of fifteen. This disorder is primarily described in young children and equally affects both sexes. Given that our symptoms started at age 4, our cases is consistent with previous research because location, gender, race, or culture do not seem to be risk factors, a spontaneous mutations of the ACVR1 gene on chromosome 4 of autosomal dominant inheritance suggests The Prescence Of A Genetics Components [14].The Potential Catastrophic , Explosive New Bone Development Causes A Biopsy Not Suitable .Lesions Biopsies , Intramuscular Injections, And Nerve Blocks ,Particulars In The Region ,Particular In The Region Of The Temporomandibular Joint, Might Cause Disease Flare-Ups [15].Recurring Cases Of Painful Swellings Of The Soft Tissues Are The Feature Of This Extremely Rare Hereditary Illness, Which Can Be Deeply Disable. Hearings Loss, Kidney Stones, And Thoracic in Sufferings Syndromes Are Among the Complications Associated With FOP. 54 FOP Patients Were Questioned for A 1999 Study Which Revealed That 52% Of Them Had Hearing Impairment [16]. A Non-Inflammatory Condition Known as Fibrodysplasia Ossificans Progressive (FOP) Is Characterized by Heterotopic Bone Growth Following Damage to The Skeletal Muscle, Tendons, Ligaments, And Fascia. Through A Global Prevalence Of 1 Case Per 2 million People, FOP Is an Incredibly Rare Disorder. There Is No Racial or Gender Bias in It. The Condition Manifests Throughout the First Ten Years of Life After Birth. Activin Receptor 1a/ Activin Kinase 2 (ACVR1/ALK2) Mutation Is the Genetic Cause Of FOP; However, Most Causes Occur Sporadically Due to Mutations. The Genetic Inheritance Pattern Is Autosomal Dominant Can Be Inherited from The Either Parent. FOP Causes Ectopic Osseous Growths in Muscles and Connective Tissues with Episodic Flare-Ups and Restricts Movements at The Sites of Involvement. Clinical Professionals Frequently Misunderstands Aggressive Juvenile Fibromatosis, A Condition Known as Lymphedema, Or Soft-Tissue Sarcoma in FOP [17]. 87%Errors Have Been Identified; Malignancy Is the Most Common Incorrect Diagnosis. Usually, Iatrogenic Damage Occurs and The Natural History Is Altered.[7]
CONCLUSION:
Stone Man Syndrome, or Fibrodysplasia Ossificans Progressiva (FOP), is a rare genetic disorder that profoundly impacts the lives of those affected, often starting in childhood. Advances in medical research have provided valuable insights into the role of the ACVR1 gene mutation in driving abnormal ossification. These discoveries have opened doors to targeted therapies, such as investigational drugs that inhibit bone morphogenetic proteins (BMPs) and gene-based treatments. Despite these advancements, significant challenges remain, including the lack of a definitive cure and the limited availability of effective therapies. Raising awareness about FOP is essential to enable early diagnosis and prevent harmful medical interventions, such as unnecessary surgeries, which can worsen the condition. Current management strategies focus on symptoms control through non-steroidal anti-inflammatory drugs (NSAIDs), bisphosphonates, corticosteroids, and supportive therapies like physical therapy. Emerging treatments in clinical trials offer hope for slowing disease progression and minimizing complications. Through continued innovation, collaboration, and empathetic care, we can aim not only to alleviate the burden of this disease but also to improve the quality of life++++++++++22. Stone Man Syndrome serves as a powerful reminder of the importance of scientific discovery and compassionate healthcare in tackling rare and devastating conditions.
REFERENCES


Sidhartha Lolla*, Lukalapu Mothilal, Uppalapu Akhila Sree, Maddineni Bhagya, Achagouni Akhilagoud, Mangalaram Sneha, A Review on Fibrodysplasia Ossificans Progressiva- Stone Man Syndrome, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 1, 969-976. https://doi.org/10.5281/zenodo.14637602
 10.5281/zenodo.14637602
10.5281/zenodo.14637602
