Department of Quality Assurance, Shri D. D Vispute College of Pharmacy and Research Center, New Panvel.
In the process of finding new drugs, analytical development and validation are crucial. It is a continuous and inter-dependent task linked with research and development, quality assurance and quality control department. Analytical method development and validation helps to demonstrate that the technique developed it precise, accurate, linear, and robust, as well as specific to analyse a drug in pharmaceutical industry. The essential part of a study is developing the analytical methodology. Advancement in analytical instruments lead to recent development in analytical methods. The excipients, active pharmaceutical ingredient (API), degradation products, residual solvents and other related substances are developed and validated using analytical techniques. To provide accurate results, analytical method validation used. This review is focusing on the important concepts, criteria, strategies, and importance of developing analytical method and validating the developed method.
In analytical method validation, sample is analysed for its chemical makeup. Analytical chemistry is of great importance. The main concern is finding the qualitative and quantitative makeup is the main concern. To identify atomic and molecular species or functional groups in a sample, qualitative approach is used, but to know the proportional of these elements, quantitative approach is used. Analytical chemistry studies how to analyse a sample to find out its chemical makeup. Finding the materials qualitative and quantitative makeup is its main concern. The functional group’s atomic or molecular structure can be obtained by a qualitative approach. On the other hand, a quantitative approach offers numerical data regarding the proportion of one or more of these elements. It is a subdivision of science to study the structure, chemical makeup and behaviour of a matter. To comprehend the identity, configuration of chemical entities, concentration. It is used in drug development processes, ranging from quality control, safety, marketing, storage. A dosage form developed for human consumption should be of highest purity without harmful impurities. Analytical techniques are used to analyse such dosage forms, since it could have a harmful effect upon consumption. Analytical methods are foundation for analytical chemistry. In order to analyse samples various analytical techniques are developed and validated [1]. Other scientific fields which require analytical data are zoology, biology, arts, space exploration, archaeology. Apart from medical diagnosis, other fields using analytical chemistry are biological, clinical research, pollutant management, applied sciences, geological testing [2]. Analytical chemistry also consists of evaluating natural and artificial materials containing more than one or one component and separating, measuring and identifying them for any chemical additives present. Qualitative and quantitative evaluation are two primary classes of analytical chemistry.
Analytical Method
A predefined detailed procedure to analyse one or more than one analytes in quantitative, qualitative or structural way is known as analytical method [3].
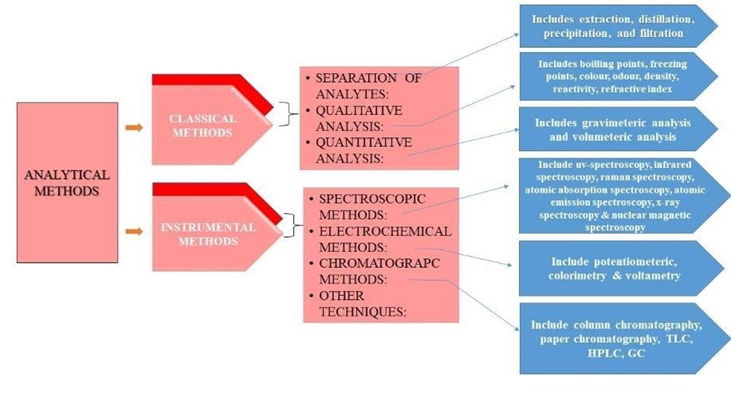
Fig 1: Analytical Methods
Typical techniques and instrumentation in analytical chemistry
In 1860 caesium (Cs) and rubidium (Rb) were discovered, Robert Bunsen and Gustav Kirchhoff invented Flame emissive spectrometry and it is the first instrumental analysis done.6 The properties of the analyte to be analyzed determine the classification of the instrumental techniques used for chemical analysis. such as solubility, specific gravity, density, transparency, turbidity, colour, freezing point, boiling point, moisture content can also be determined. Both physical and chemical changes are brought by chemical reactions [6] [7] [8].
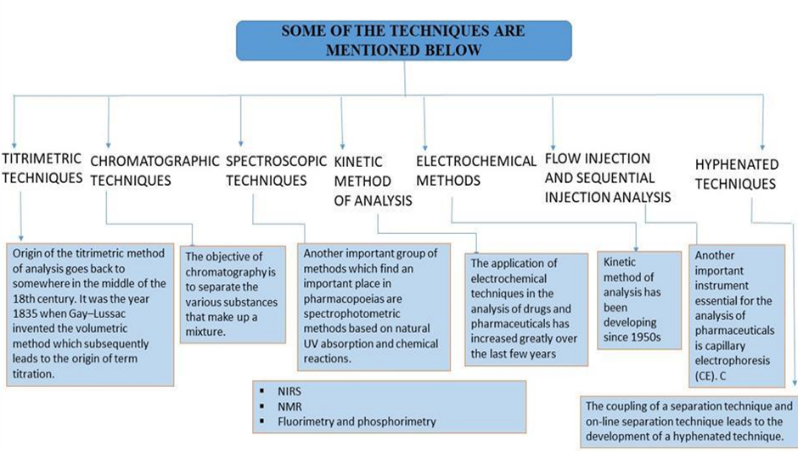
Fig 2: Techniques and Instrumentation in Analytical Methods.
Fundamentals Of Spectroscopy
It is defined as a scientific term to study the interaction between matter. Radiation is emitted during absorption or emission of energy by matter. Emission and absorption are two kinds of spectroscopy. The electromagnetic radiation absorbed by the sample are studied using absorption spectroscopy such as UV-visible, infrared, microwave, nuclear magnetic resonance, radio wave spectroscopy. Flame photometry, fluorimetry, emission spectroscopy are techniques used to analyse electromagnetic radiation emitted by a sample. Various samples are studies for their atomic and molecular structure using spectroscopy. Atomic spectroscopy includes atomic absorption and flame photometry. It explains how electromagnetic radiation interacts with atoms thereby producing energy fluctuations at atomic level. Molecular spectroscopy includes ultraviolet and infrared region of spectroscopy; it explains how radiation interact with molecules to cause a change in their energy. [4]
Ultraviolet-visible spectrophotometry
The most frequently used technique in pharmaceutical analysis is UV-visible spectroscopy. It is employed to quantify the quantity of visible or ultraviolet light that the compound in a solvent absorbs. It calculates the function or ratio of two UV-visible light beams' intensities. A spectrophotometer is used to qualitatively analyse and identify organic substances and to measure the amount of radiation absorbing species, qualitative spectrometry can be used if any existing data is available. Small amount of samples can be analysed in spectrophotometric analysis, it is easy to use, time saving and specific. Beer and lambert’s law governs the principles involved in spectrophotometry. [5]
Table 1: Regions of Electromagnetic Spectrum
|
Region |
Wavelength |
|
Far (or vacuum) ultraviolet |
10-200nm |
|
Near ultraviolet |
200-400nm |
|
Visible |
400-750nm |
|
Near infrared |
0.75-2.2µm |
|
Mid infrared |
2.5-50µm |
|
Far infrared |
50-1000µm |
UV Method Development
Everyday an increasing number of new medications are introduced into the market. A delay occurs in the drug commercialisation and when it is included in the official pharmacopoeias, this results in flaws in current usage, recently reported toxicities, patients developing resistance, expanded usage and competitors developing advanced medicines. In these circumstances, pharmacopoeias will not have scientific methods for such medicines. This calls to develop a new theoretical framework for such medications. Quality is an essential element in all products and services, since drugs deal with people’s lives [9].
The normal values for a medicine are evaluated using official analytical methods instead of using the administrative systematic methods, it is suggested to use logical methodology, an important step when developing new pharmaceutical medicine is security testing [10].
Degradation products produced and dynamic medication test are used to determine the usability of pharmaceutical items that are to be used in realistic timeline. The following are some explanations for the advancement in drug analysis techniques:
When any drug or combination of drug is not included by any pharmacopoeias as official, following could be the causes:
Various techniques for UV spectrophotometric multicomponent analysis.
Instrumentation of UV-Visible spectroscopy
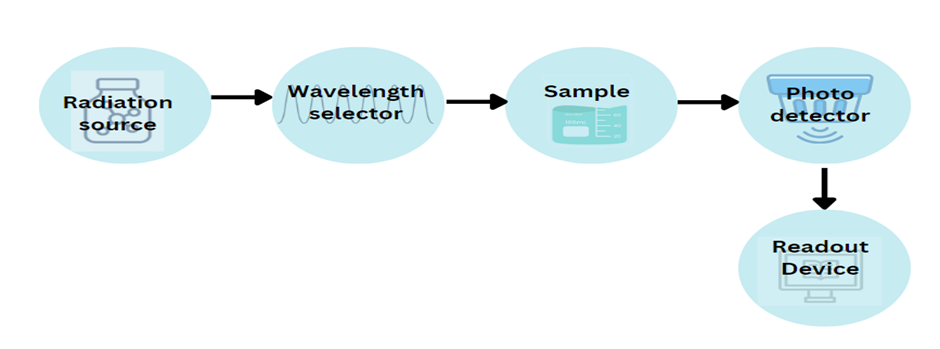
Fig 3: Instrumentation of UV-Visible spectroscopy
Radiation source: Tungsten, deuterium discharge, hydrogen discharge, xenon discharge, and mercury lamps are commonly used as the radiation sources. (figure 3)
Introduction To Chromatography
Separating mixtures by physicochemical technique is chromatography. It is a technique that separates mixture of substances into its component parts using both, mobile phase and stationary phase [14] [15].
HPLC
The most popular tool in analytical chemistry is HPLC (high performance liquid chromatography). It is used in distinguishing, recognize, and measure the liquid soluble chemicals. The most accurate analytical techniques quantitative and qualitative spectroscopy are frequently applied to drug product analysis [16]. HPLC is a separation technique in which a minor quantity of liquid sample volume is injected into a tube packed with small particles, referred to as the stationary phase, that are 3 to 5 microns in diameter. Liquid (mobile phase) is injected with the help of high pressure provided by pump, which moves individual components of sample down the column packing. Column packing has different chemical or physical interactions within the molecules and packed particles, keeping the components separated from each other.
Instrumentation of HPLC
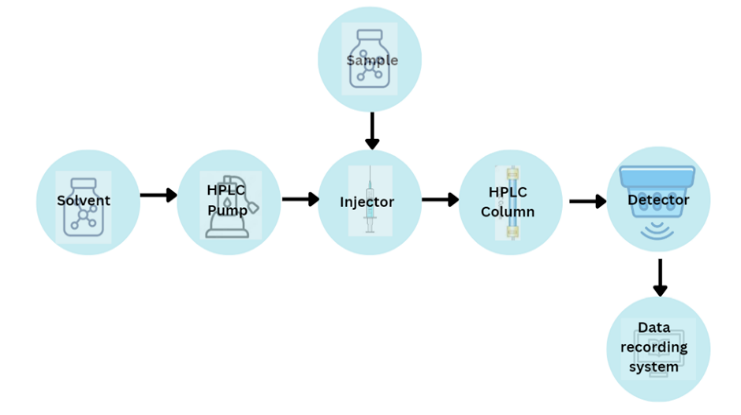
Fig 4: Instrumentation of HPLC
Mixing system, solvent reservoir and degassing system:
The solvent reservoir is used to store the mobile phase. These containers are compound stainless steel or glass. The mostly used solvent reservoirs are made of glass [17]. The pump also needs to combine solvents with extreme precision and accuracy along with distributing the mobile phase. Low pressure mixing and high pressure mixing are the two primary types of mixing units [18]. The trapped air bubbles are eliminated from the solvent using the degassing system. Ultra-sonication and filtration are the degasser procedures which assist with the degassing.
2. High pressure pump:
A liquid is pushed with the help of pump to provide a particular flow rate. Millilitres per minute (ml/min) is the unit for measurement of flow rate [19]. Pump pressure typically ranges from 400 to 600 bar (6000 to 9000 psi), and 1-2 ml/min is the normal flow rate.
Mobile phase is sucked by the pump from the reservoir and is forced into the column and then is passed to the detector. The operating pressure is 40000 KPa. Particle size, flow rate, column dimensions, and mobile phase composition are a few operating parameters [20].
3. The sample injector:
The sample injector is used to introduce the sample liquid into the mobile phase. The sample valve is positioned between the pump and the column. An injector, also called an auto sampler, is used to inject the sample into the continuously running mobile phase that feeds the sample onto the HPLC column. The sample quantities range from 5 to 20 microliters (µl) [21]. Automatic and manual are two different types of injectors.
4. Column:
The actual components separation occurs in the column. Columns are made of stainless steel and length is ranging from 5 to 25 cm with its internal diameter is 2 to 4 cm.
5. The detector:
Each component that elutes from the column is identified by the detector, which transforms the data into an electrical signal. Bulk property detectors and specialized detectors are the two types of detectors that are employed. UV-VIS, fluorescence, spectrometric detectors and photo diode array are examples of specific detectors. Refractive index, light scattering and electrochemical detectors are examples of bulk property detectors.
6. Data recorder
The output is captured as a series of peaks, and a computer is used to automatically compute the data by determining the area beneath each peak.
Developing Analytical Method
The process to develop analytical procedure which is accurate, to determine the composition of a formulation is known as analytical method development. Analytical method development is the process of demonstrating that an analytical technique is suitable for use in a laboratory. Protocols should be followed while developing analytical methods meeting acceptance criteria according to the ICH guidelines and utilized only in GMP and GLP environments. Q2 (R1) [24].
Techniques, method, procedure, protocol and the steps of analytical method development
Criteria to develop new analytical method
The foundation for determining the product is drug analysis. Due to a significant delay between a drug's release date in the market and the date it is included to pharmacopeia, it causes potential ambiguities in ongoing and increased use of such medications. Reporting of new toxicities, patients develop resistance, launching of more effective medications by the competitors pose a major concern while developing an efficient analytical method [22]. There could be a possibility that no pharmacopoeia has accepted the new medication or medication combination because of patent regulations, a proper analytical process might not be published in the literature for the medicine and analytical techniques might not be accessible in the form of formulation excipients. There might not have been development of analytical techniques for a medication when used with other medications. It is possible that the current analytical techniques require costly solvents and reagents. It could include laborious extraction and separation techniques, which might not be dependable [23].
Necessity to develop a method
The drug evaluation includes identification, characterisation, and resolution of the medications in combination, such as dosage forms and in organic fluids. The goal of developing analytical method is to produce data regarding efficiency (which may be closely linked to the requirement for a specific dosage), impurity (safety of the medication), stability (identifies breakdown product), bioavailability (includes crystal type, drug homogeneity, and medication release), and manufacturing impact parameters to confirm that the medicinal product being produced is steady.
Fig 5: Steps involved in method development
Need to develop analytical method and its validation
The requirement to develop and validate analytical technique arose from global rivalry, keeping the standards of highly valuable items in the commercial domains, market domains, and ethical considerations. A few of the well-known groups are in charge of the quality standards:
Validation
FDA (food and drug administration) states validation as a process and production technique intended to guarantee the uniqueness, excellence, potency, and purity of the pharmaceutical products. to demonstrate that the procedure and the system fulfil the necessary specifications. The concept of validation was developed in 1978 in United States. Over the years, the concept of validation has broadened to encompass a variety of activities, such as computerized systems for clinical trials, labelling or process control, and analytical methods to guarantee the quality of medical products and therapeutic substances. It is advised to think validation as an essential component of cGMP. It refers to the assessment of validity or the process of demonstrating efficacy. Validation is a collaborative effort which involves individuals from several plant branches. A higher level of certainty that the equipment or product will meet the goals of the analytical application is provided by the "process of establishing documented evidence," sometimes referred to as "method validation." [25] [26] [27]
According to USP: Analytical method
According to ICH method
"Establishing a documented evidence, which provides a high degree of assurance that a specific activity will consistently produce desired result or product meeting its predetermined specifications and quality characteristics" is defined as validation by the International Committee of Harmonization. For the trace impurity assay, a distinct methodology and set of acceptance standards are required than for the major component assay. Some aspects of the HPLC process may be significant because of differences in the backgrounds and abilities of employees, as well as in the HPLC instrumentation, lab supplies, and reagents. The methods may need to be adaptable in order to create different forms of the same medication with different potencies or physical characteristics. System appropriateness, linearity, accuracy, precision, specificity, robustness, limit of detection, limit of quantification, and stability of samples, reagents, and equipment are all important factors in method validation study [28].
Method validation parameters
1. Accuracy:
Accuracy is the degree to which the measured value closely resembles the true value. To match the true value, a sample with a known true value is analyzed using a highly accurate method. Recovery studies are determined by accuracy.
2. Precision:
It is an indicator of how closely different values from the same sample are related. RSD (relative standard deviation) is the way it is expressed. To obtain precise results under explanatory conditions, several homogenous samples are tested.
%RSD = Standard deviation/Mean ×100
According to the ICH Guidelines, repeatability must appropriately conform to at least nine determinations within a specified range or a minimum of six determinations at 100 % of the test concentration.
3. Linearity:
The potential to obtain a linear response which is directly proportional to concentration of the sample in solution. Expressed as confidence limit around the slope of the regression line.
4. limit of detection (LOD):
The lowest amount of analyte in a sample that can be detected but not quantified. It is expressed as concentration at specific signal to noise ratio. LOD= 3.3 × S/SD
5. limit of quantitation (LOQ):
Lowest concentration of analyte that can be quantified is known as limit of detection. Signal to noise ratio of 10:1 is recommended by ICH.
LOQ= 10 × S/SD
6. Specificity:
The ability to provide interference free signals is one of the notable feature of HPLC. The capability of analytical technique and distinguish and detect the analyte in mixture is referred to as specificity according to ICH is defined as the ability to evaluate the analyte in the presence of several substances that may potentially by present. These can be contaminants, matrix, degradants and other things. The following implications follow from the definition:
7. Range: It refers to the range of the analyte levels that have been determined with appropriate accuracy, precision and linearity, and the lower level. It is calculated by linear response curve or non-linear response curve. Unit is same as in test findings.
8. Robustness: Ability to remain unchanged due to minor, intentional alterations in parameters such as column temperature, flow rate, pH, composition, or mobile phase is termed as robustness [29] [30]
Summary Of Literature Survey Of Different Drug.
Table No: 2 Summary of literature survey of different drugs
|
Sr.no. |
Drug |
Instrument |
Column |
Flow rate |
Mobile phase |
Retention time |
Linearity |
Ref
|
|
1 |
Ivabradine Hydrochloride |
HPLC |
C18 (250×4.6mm;5µm) |
0.8 mL/min |
Phosphate buffer: Methanol (60:40 v/v) |
6.55±0.05 min |
r2=0.9998 |
[31] |
|
|
|
HPLC |
C18 (250mm×4.6mm;5µm) |
1 mL/min |
Methanol: ACN (80:20: v/v) |
5.8 ml/min |
r2=0.998 |
[32] |
|
|
|
HPLC |
ODS-3V (250mm×4.6mm) |
0.7 mL/min |
0.5% Formic acid (pH 7.0): Acetonitrile (65:35 v/v) |
8.67±0.0376 |
r2=0.999 |
[33] |
|
2 |
Brivaracetam |
HPLC |
Agilant column (4.6×150mm; 5µm) |
1.0 mL/min |
Phosphate buffer: Methanol (25:75 v/v) |
2.182 Min |
r2=0.992 |
[34] |
|
|
|
RP HPLC |
ODS RPC18,5mm, 15mm x 4.6mm |
1.0 mL/min |
phosphate buffer: methanol: acetonitrile (30:35:35% v/v) |
2.183 min |
r2=0.999 |
[35] |
|
3 |
Pregabalin |
HPLC |
Hypersil BDS, C8, (150×4.6mm, 5µm) |
1 mL/min |
Phosphate buffer (pH 6.9): ACN (95:05) |
*** |
r2=0.9998 |
[36] |
|
|
|
HPLC |
C18 Column (250×4.6mm; 3.5 µm) |
0.8 mL/min |
Acetonitrile: Methanol (80:20) |
*** |
r2=0.999 |
[37] |
|
|
|
HPLC |
Inertsil ODS 3V, (250× 4.6mm ID, 5µm) |
1 mL/min |
Phosphate buffer: Acetonitrile: Methanol (80:10:10) |
4.7 min |
r2=0.999 |
[38] |
|
4 |
Levetericetam |
HPLC |
Prontosil C18 Column (150×4.6 mm; 5µm) |
1.2 mL/min |
Buffer solution (pH 2.8): Acetonitrile (90:10) |
4 min |
r2=0.9999 |
[39] |
|
|
|
UV |
- |
- |
H2O |
*** |
r2=0.9982 |
[40] |
|
|
|
HPLC |
Flexit C18 Column (150×4.6mm; 5µm) |
1 mL/min |
Water: Methanol (75:25) |
*** |
r2=0.9989 |
[41] |
|
5. |
Lamotrigine |
LCMS |
Chromolith, C18 (50×4.6mm; 4µm) |
0.500 mL/min |
Phosphate buffer (pH2.5) Acetonitrile (80:20) |
*** |
r2=0.9987 |
[42] |
|
|
|
HPLC |
Intersil C8 column (250mm length, 4,6mm) |
0.8 mL/min to 1.2 mL/min |
ACN: Methanol (60:40 v/v) |
2.542 min |
r2=0.9905 |
[43] |
|
|
|
HPLC |
C8 (4.6mm ID×150mm. 3.5µm, Make:XTerra) |
0.8 mL/min |
Acetonitrile: potassium dihydrogen phosphate buffer of pH= 7.0 (60:40) |
2.797 min |
r2=0.998 |
[44] |
|
6. |
Ketoprofen |
HPLC |
C18 Column (5µm) |
1.0 mL/min |
Acetonitrile: Milli Q water acidified by 0.1 % (v/v) Formic acid |
3.06 min |
r2=0.9997 |
[46] |
|
|
|
UV |
|
|
NaHCO3 |
|
r2=0.998 |
[47] |
|
|
|
HPLC |
C18 Column, 5µm (25×4.6 mm |
1mL/min |
Methanol: Water (70:30) |
<10> |
r2=0.9999 |
[48] |
|
7. |
Omeprazole pellets |
Ultra fast gradient LC method |
Column (50×20mm id) |
1.0 mL/min |
0.15% (v/v) trifluoracetic acid (TFA) in water & 0.15% (v/v) TFA in acetonitrile |
0.74 min |
r2=0.998 |
[50] |
|
|
|
RP HPLC |
RP?C18 column |
1.5 mL·min?1 |
Phosphate buffer (pH 7.4) and acetonitrile (70:30 v/v) |
5 min |
r2=0.9995 |
[51] |
|
|
|
LC-MS/MS
|
RP-C8 (50 X 4.6mm, 5µm particle size) |
0.6 mL min-1 |
Methanol: water (containing 0.5% Formic acid) 8:2
|
*** |
r2=0.9999 |
[54] |
Based on the literature survey of various analytical methods for different pharmaceutical compounds, I have selected Ketoprofen and Omeprazole pellets for the current study. analytical methods used for estimating ketoprofen and omeprazole in pharmaceutical formulations. Based on your literature survey, it seems there is limited research available on the use of UV, HPLC, HPTLC, and stability-indicating RP-HPLC specifically for the combination of ketoprofen and omeprazole. This observation could indicate a gap in the existing literature, and it may provide a unique opportunity to explore and develop an analytical method for this combination.
The following outlines the detailed breakdown of the research work.
Ketoprofen and Omeprazole
Ketoprofen is a nonsteroidal anti-inflammatory drug (NSAID), commonly used for pain relief and inflammation.
Omeprazole is a proton pump inhibitor (PPI) used to treat gastroesophageal reflux disease (GERD) and other acid-related disorders.
Since these two drugs often appear together in combination formulations (to reduce the gastrointestinal irritation caused by NSAIDs), developing a reliable and robust analytical method for their simultaneous estimation in pharmaceutical dosage forms would be highly valuable.
Existing Literature Insights
UV Spectroscopy: UV-Vis spectrophotometry is a common method for drug quantification but has limitations in terms of selectivity and sensitivity, especially for combinations. There is a need to explore whether the wavelengths used for each drug could offer a distinct signature for ketoprofen and omeprazole.
HPLC: High-performance liquid chromatography (HPLC) is widely used for the separation and quantification of drug mixtures. The literature suggests that there is some work done on estimating ketoprofen and omeprazole individually or in combination with other drugs, but possibly with some method limitations in terms of sensitivity, selectivity, or reproducibility.
HPTLC: High-performance thin-layer chromatography (HPTLC) might be less common in this specific context but could be explored as a potential technique due to its lower operational costs and ease of use in pharmaceutical analysis.
Stability-Indicating RP-HPLC: Stability-indicating methods are essential for assessing the stability of drugs under various conditions (like temperature, humidity, and light). Since ketoprofen and omeprazole might undergo degradation, developing a stability-indicating RP-HPLC method for the simultaneous estimation of these drugs could be a significant contribution.
Potential Research Directions
Development of a Novel Analytical Method:
Work could be done on optimizing an RP-HPLC method that can separate and quantify both ketoprofen and omeprazole simultaneously. You may need to fine-tune the mobile phase composition, flow rate, and column type to achieve the required resolution and sensitivity.
Stability-Indicating Method:
Develop a stability-indicating RP-HPLC method for the combination drug. This would involve subjecting the drug to stress conditions (e.g., heat, light, oxidation) and ensuring that the method can distinguish the drug from its degradation products.
Validation:
To ensure that the method developed adheres to validation criteria (e.g., specificity, accuracy, precision, linearity, limit of detection, limit of quantification, robustness) to ensure its suitability for routine analysis.
Exploring Analytical Alternatives:
Explore the possibility of using other techniques, like HPTLC, for simpler and more cost-effective analysis. Although HPTLC may not be as sensitive as HPLC, it might be suitable for routine quality control if combined with suitable detection systems (e.g., densitometry).
Table 3: Summary of literature survey of Ketoprofen and Omeprazole pellets
|
Sr.no. |
Drug |
Instrument |
Column |
Flow rate |
Mobile phase |
RT |
Linearity |
Ref
|
|
1 |
Ketoprofen |
HPLC |
C18 Column (5µm) |
1.0 mL/min |
Acetonitrile: Milli Q water acidified by 0.1 % (v/v) Formic acid |
3.06 min |
r2=0.9997 |
[45] |
|
|
|
UV |
|
|
NaHCO3 |
|
r2=0.998 |
[46] |
|
|
|
HPLC |
C18 Column, 5µm (25×4.6 mm) |
1mL/min |
Methanol: Water (70:30) |
<10> |
r2=0.9999 |
[47] |
|
|
|
HPLC |
Shim-pack column vp-ODS (250×4.6 mm) |
1mL/min |
Ethanol: Phosphate buffer (80:20 v/v) |
10 min |
r2=0.9999 |
[48] |
|
|
|
RP-HPLC |
C18 Type MG column (250×4.6mm) |
1.0 mL/min |
Acetonitrile: 0.02 M Potassium dihydrogen orthophosphate buffer (40:60) |
*** |
r2=0.999 |
[49] |
|
2 |
Omeprazole Pellets |
Ultra fast gradient LC method |
Column (50×20mm id) |
1.0 mL/min |
0.15% (v/v) trifluoracetic acid (TFA) in water & 0.15% (v/v) TFA in acetonitrile |
0.74 min |
r2=0.998 |
[50] |
|
|
|
RP-HPLC |
RP?C18 column |
1.5 mL·min?1 |
Phosphate buffer (pH 7.4) and acetonitrile (70:30 v/v) |
5 min |
r2=0.9995 |
[51] |
|
|
|
RP-HPLC |
C18 (150×4.6mm; 5µm) |
0.5mL/min |
Acetonitrile: Phosphate buffer solution (60:40 v/v) |
7.71 min |
r2=0.9988 |
[52] |
|
|
|
RP-HPLC |
C18, (250 x 4.6 mm, 5µ) |
1.0 mL/min |
Phosphate buffer (pH 7.4) and acetonitrile in the ratio of 60:40, v/v |
7.71 min |
r2=1.0000 |
[53] |
|
|
|
LC-MS/MS |
RP-C8 (50 X 4.6mm, 5µm particle size) |
0.6 mL min-1 |
Methanol: water (containing 0.5% Formic acid) 8:2 |
- |
- |
[54] |
|
3 |
Ketoprofen + Omeprazole pellets |
HPLC |
Phenomenex Luna C18 (2) column (150 × 4.6 mm, 5 ?m) |
0.8 mL/min |
Mobile phase A consisted of 0.01 M KH2- PO4 and 3 mL of TEA (pH 7.0 adjusted with H3PO4). Mobile phase B contained a mixture of Milli Q water and ACN in the ratio of 10:90 (v/v). |
3.9 min. |
r2=>0.999 |
[55] |
|
|
|
HPLC |
LiChrosorb C18, 125 mm x 4.6 mm, 5 ?m |
1.5 ml/min. |
Phosphate buffer (pH adjudted to 4.6 with ortho-phosphoric acid) and methanol (25:75v/v) |
3.05 and 6.96 min |
Ketoprofen- r2=0.9999 Omeprazole- r2=0.9998 |
[56] |
|
|
|
HPTLC |
- |
- |
Chloroform: methanol 9:1 (v/v) |
- |
Ketoprofen- r2=0.999 Omeprazole- r2=0.999 |
[57] |
CONCLUSION
In conclusion, this review outlines essential guidance on method development, validation, and its critical role in pharmaceutical analysis. It highlights the importance of detailing various validation types, and executing precise methods to ensure their effectiveness for intended applications. The primary goals of analytical technique development identification. The primary goals of analytical technique development identification, purification, and qualification are fundamental in verifying that pharmaceutical products meet quality standards. Ensuring the reliability and accuracy of methods in the pharmaceutical industry is crucial, as it supports the production of high-quality medicines and materials, ultimately protecting consumer health and safety.
REFERENCES





Mukesh Patil*, Roshani Dhumal, Ashish Jain, Srushti Kadave, Aishwarya Patil, A Review: Analytical Method Development and Validation, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 2350-2365. https://doi.org/10.5281/zenodo.14506358
 10.5281/zenodo.14506358
10.5281/zenodo.14506358
