Department of Pharmaceutics, The Oxford College of Pharmacy, Bangalore, Karnataka 560068.
Pre-formulation studies are a crucial phase in the development of pharmaceutical dosage forms serving as the foundation for an effective formulation design. Pre-formulation studies select and characterize excipients according to their functionality, compatibility with APIs, and influence on formulation properties. They are conducted to assess the mechanical and physical properties of dosage forms in order to ensure patient compliance and proper drug delivery. Compaction studies, powder flow characteristics, and particle size analysis help to improve the uniformity and reproducibility of dosage forms and optimize the formulation process. Furthermore, pre-formulation studies evaluate formulation stability in both accelerated and long-term storage conditions, helping to determine the best packaging options and storage conditions.
The development of pre-formulation occurred in late 1950s and around 1960s due to change in the focus of industrial pharmaceutical products. When developing a new drug substance, the synthetic chemist may record particulars on their own or in collaboration with experts from other fields, like pre-formulation specialists. It is suitable to classify as pre-formulation data. Knowledge about drug characters, potency in relation to competing goods, and dose is necessary before beginning pre-formulation investigations, a search of the literature for stability and decay data, the suggested drug administration route, and information about formulation techniques. (1) It is defined as “the examination of the physical and chemical properties of a medicinal ingredient, both alone and in combination with excipients.” (2) These are intended to impart all relevant information, particularly about the physiochemical, physio-mechanical and pharmaceutical characteristics of excipients, packaging materials and API (3) This helps in determining the potential of drug molecules. Thus, it can be regarded as an essential aid for decision-making in the phases of drug development and discovery. These studies are usually performed before formulation of different dosage forms. Some of the factors including stability, dissolution rate, drug solubility, polymorphic forms, partition coefficient are important in pre-formulation studies. (4)
Need for dosage forms
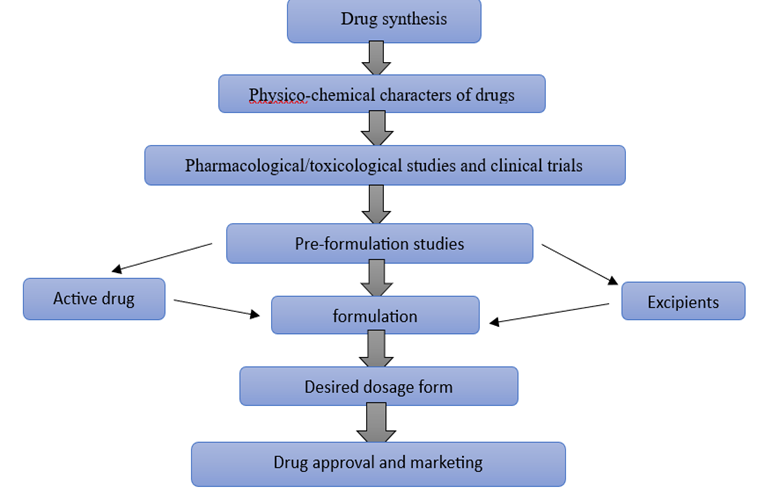
Figure 1: Steps involved in drug formulation and their approval to market.
Physicochemical parameters:
C. Solubility studies
D. Stability studies
1.Organoleptic properties:
Colour: The colour of the drug should be eye appealing that can be determined either by visible or instrumental methods and varies from batch to batch. Preliminary batch recording and specification creation are highly helpful for later manufacturing. The body can be covered in a different colour if this is deemed undesirable (5)
Odor and taste: When using less soluble chemical forms of drugs, they can be made more appealing by adding coatings, flavours, or excipients. Precautions are taken while handling drug compounds that can cause skin irritation. Stability and bioavailability may be impacted by the flavours, colours, and excipients utilized (5)
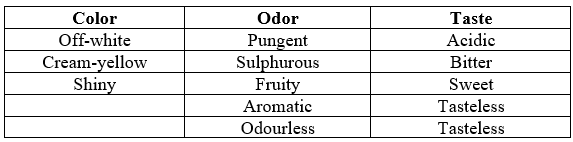
Table1: Excipients like colour, Odor, and taste of drug
Bulk characterization:
They are important to analyse fixed form that could result from synthetics stages, like the existence of polymorphs. During development phase, bulk parameters including size of the particle, surface morphology, bulk density can be changed to prevent making inaccurate solubility and stability predictions that rely on certain crystalline forms.
a) Crystallinity and polymorphism:
Crystalline: Crystallinity is referred to as the structure of solid compounds and they disappear in vapor and liquid states. Drug’s physicochemical properties, such as chemical stability and flow ability, can be affected by the crystal habit and internal structure. The internal structures can be characterised as cubic, tetragonal, hexagonal, rhombic, etc., and solid structure as tubular, bladed, needle, platy and prismatic. Modifying internal structure also modifies crystal habit (6)
Amorphous: Atoms or molecules are arranged randomly as liquid in this form.
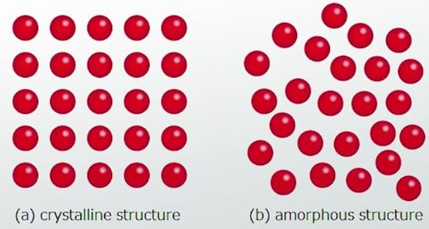
Figure2: Morphology of crystalline and amorphous structure
Polymorphism:
The term "polymorphism" refers to the phenomenon of a drug which can occur in several crystalline form is referred to polymorphs. "Hydrates" is the term used when the incorporated solvent is water. Anhydrous compounds are defined as those that have no water in their crystal structure. (6) There are 2 types of polymorphism:
1) Enantiotrophs: Enantiotrophs are those that may undergo reversible transformations into other forms through changes in pressure or temperature ex: Carbon
2)Monotrophs: This transition occurs only in one direction and is called monotropic polymorphism ex: chloramphenicol palmitate
Pseudo polymorphism: Pseudo means false. Hot stage microscopy can be used to distinguish between pseudo polymorphs and real polymorphs by analysing the melting behaviour of silicon oil.
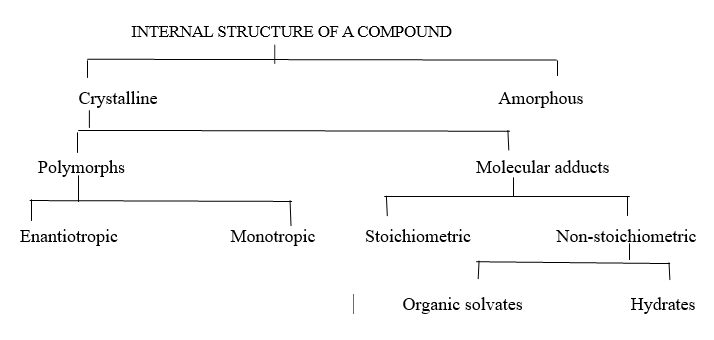
Figure3: Classification based on internal structure and crystal habit
b) Hygroscopicity [the ability to see in low light]
A solid's ability to absorb moisture from its surroundings is known as its hygroscopicity. Moisture and water vapour can affect a wide range of chemicals and salts. When substances come into contact with moisture, they can absorb it through surface or bulk adsorption, capillary condensation, chemical reactions, precipitation of a solution. A solid undergoes deliquescence when thin layer of water on the surface dissolves and becomes saturated. It has been demonstrated that the liquid film surrounding the solid becomes saturated when a certain threshold relative humidity is reached and moisture is absorbed to the point where crystallization occurs. It can be characterised by gas chromatography, gravimetric method and Karl Fischer method. (7)
Note: While dry material may hydrate and become less soluble, amorphous compounds may absorb water and re-crystallize. Additionally, the weight change caused by sorption may result in potency errors.

Table2: classification on basis of moisture absorbance by the materia
c)Densities: It is the mass to volume ratio.
1. Bulk density:
The process involves figuring out how much powder, with a known mass, passed through the screen.
a) Compound bulk densities differ significantly based on the grinding, compounding, or crystallisation process used.
The characteristics of powder flow can be influenced, which in turn affects the uniformity of low dose formulations when there are notable discrepancies in the densities of excipients and active pharmaceutical ingredients. Additionally, it can also impact the size of high dose capsule products. (8)
Bulk Density = Mass of sample/Volume
2. Tapped density
It can be measured after taking sample in a measuring cylinder and tapping it on a slab provided its opening is closed with hand or using any other cover. (9)
Tapped Density = Mass of sample/Tapped Density Volume
3. True density: Total sample mass to the volume, volume of the voids should be eliminated.
d)Powder flow properties
For tableting processes to be effective, powder flow characteristics are crucial. Therefore, a drug's mobility should be taken into account during pre-formulation review, particularly if a high dose is anticipated. Powders can either flow freely or cohesively (not freely). Flow characteristics are affected by differences in adsorbed moisture, static charge, particle size, density, and form. (10)
Flow property can be characterised by
Carr’s Index
Carr’s index = [Tapped density - bulk density / tap density] 100
Hausner ratio
Hausner’s ratio = Tapped density/Bulk density
Angle of repose

Table 3: represents type of powder flow based on the values of the angle of repose
Formula for calculating angle
? = tan-h/r
? = angle of repose
h= heap’s height
r = heap’s radius
Method to determine angle of repose: (Fixed funnel, free- standing cone)
The funnel is slowly filled with powder, and the tip is closed with a finger to prevent the powder from flowing too fast. The heap's peak is formed when powder passes through a funnel that is fixed at a specific height and reaches tip of the funnel. The digression of the angle of repose are calculated as ratio of the powder pile's base height (h) to its mean radius (r). (11)
e) Properties of Compression.
Tiny quantities of potential drug candidates can be used to test their compression properties (elasticity, fragment ability, plasticity, and punch filming tendency). This characteristic helps in the appropriate choice of formulation ingredients.
A powder's "compact ability" is its capacity to be compacted into a tablet with a specific tensile strength, whereas its "compressibility" is its capacity to reduce in size while under pressure. With use in density measurements, it is possible to forecast the flow characteristics. (12)
f) Particle size
The finished tablet's homogeneity is also influenced by size. Mutual sieving, or unintended separation of substances in a mixture, it can happen when there are significant size variations between the active ingredients and excipients. This makes complete mixing challenging, if it can be achieved at all, to maintain during the subsequent processing steps. (12)
3) Solubility studies
a) Intrinsic solubility determination
The intrinsic solubility should be examined at two temperatures to ensure good physical stability, extend short-term storage, and preserve chemical stability until more definitive data is available and that is: 4 to 5°C. 37°C to facilitate the assessment of biopharmaceuticals.
Step 1: It is important to identify every element that influences the solubility and dissolution.
Step 2: The medication is agitated at a constant temperature and an excess is distributed throughout the medium.
Step 3: Remove the slurry samples in relation to time.
Step 4: Use centrifugation or filtering to make ampoules clear.
Step 5: To determine a plateau concentration, assay the clear samples for drug content and evaluate using UV, HPLC, GC, and other techniques. (13)
b) pKa determination
The pH distribution hypothesis is based on the sites and characteristics of absorption for various drugs. Usually, the drugs used are potentiometric titration to calculate the dissociation constant, or pKa. When a drug is given to the biologic membrane, its degree of ionization is determined by its dissociation constant (pKa) and the pH of the solution in which it is dissolved. (14)
For acidic compounds: PH = pKa + log [ionized drug] / [unionized drug] For basic compounds: PH = pKa + log [unionized drug] / [ionized drug]
c) Partition coefficient.
This ratio, which is also known as the distribution coefficient or partition coefficient are mostly unaffected by the concentration of diluted a certain solute species' solutions.
When a solute is given to a combination of two immiscible liquids, it will diffuse throughout the two phases and attain a temperature equilibrium. The ratio of the unionized drug distributed between the organic (upper phase) and aqueous (lower phase) phases at equilibrium describes the distribution of the solute (unaggregated & undissociated) between the two immiscible layers. (15)
Po/w = (C oil/water) equilibrium
Log P= (Unionised)organic / (Unionised)aqueous
d) Common Ion effects:
The addition of common ions frequently decreases the soluble nature mildly dissolvent electrolyte. The process is known as salting out and is caused by the elimination of the water molecule as the solvent as a result of competitive hydration of other ions. Common ions are a common interaction with solvent that are frequently disregarded. Temperature, pH's effect, and other variables like ionic strength are among those that influence the rates of deterioration. Presence of chelating agent, co-solvent, antioxidants, and O2 in both instances. (15)
e) Solubilization
Small-scale research should be conducted to identify potential solubilization processes in drug candidates undergoing pre-formulation that are either poorly soluble in water or insufficiently soluble in the proposed dose form of solution. Co-solvents can generally be added to aqueous solutions to increase solubility. Using appropriate cosolvents which is ethanol, glycerol, and propylene glycol can increase the solubility of poorly soluble nonelectrolytes by orders of magnitude. It is common practice to use ethanol, sorbitol, glycerin, and PEG as authorized cosolvents when creating aqueous liquids for oral solutions. Dimethylacetamide is a common ingredient in parenteral products. (16)
Techniques to increase solubility
f) Thermal Effect
When mole of solute dissolves in a significant amount of solvent, heat is either emitted or absorbed. This is known as the heat of solution. It is ascertained using the saturated solution's solubility value, which is adjusted at a certain temperature within the desired. When the temperature rises, these chemicals become more soluble. During their disintegration, exothermic compounds release heat. A rise in temperature would cause these chemicals to become less soluble. Heat has the potential to ruin medications or alter the solution in other ways, therefore caution should be used. If the solution's heat is positive, the temperature of the solution will rise, enhanced the solubility of the medication. The heat of solution for weak acids and weak bases dissolved in water that are non-electrolyte and unionized ranges from 4 to 8 Kcal/mol. (17)
4)Stability studies
a) Solution stability
When comparing to dry state, the degradation in solution phase is rapid. Here, the importance is to make sure that the drug does not degrade in presence of GI fluid. If the drug's solution stability is poor, the formulator may be compelled to select a less soluble salt form, so long as the bioavailability is not adjusted. It includes pH, oxidation, co-solvent, temperature, Ionic strength and light. (18)
b) Solid state stability
Investigation and determination of stable storage conditions for drugs in the solid state, as well as identification of a suitable excipient for a formulation, are the main goals of this study. The chemical instability may be due to following reactions like hydrolysis, photolysis, oxidation and pyrolysis. The drug's response to any of these attacks is determined by its chemical structure. There will be some free moisture content in all solid dosage forms. This unbound water can facilitate the chemical reaction between the drug and its excipients. (19)
c)Toxicology formulation
It is recommended that these studies assess the stability and potential homogeneity issues in toxicology preparation samples. The usual methods of giving drugs to animals are through their diet, oral gagging of drug solution, or the drug suspension in aqueous medium. Water, enzymes, vitamins, moisture content and minerals (metal ions) of feed can undermine the stability of the drug and drastically reduce its shelf life. Toxicologic preparation of solution and suspension are recommended to verify the ease of manufacturing of toxicological preparation and keep it in flame-sealed ampoules at different temperatures. (19)
d)pH rate profile (20)
The extreme of pH catalyses the degradation of most drug. The most stable pH range for many drugs is 4-8. Low buffer capacity injections help prevent unnecessary disruptions to the blood's homeostatic pH.
Stability will increase with more water miscible solvents in the formulation
ex: Chlordiazepoxide injection with 20% propylene glycol
e) Compatibility
Understanding how drug excipients interact will help the formulation choose the right excipients. To optimize the possibility of masking an interaction, the pre-formulation screening method for drug-excipient interaction presented here only needs 5 mg of the drug in a 50% combination with the excipients. For optimal oxidation and paralytic effect at standard heating rates on DSC, mixtures should be analysed in nitrogen, encompassing range of temperatures to account for any variations in temperature brought on by the medication as well as appearance or disappearance. Drug excipient combination thermograms with one or more peaks are thought to be an indicator of an interaction. (20)
CONCLUSION
Pre-formulation studies serve as a fundamental stage in pharmaceutical development, guiding the formulation process for optimal drug design. By systematically assessing physicochemical properties, researchers gain insights into potential challenges and avenues for improvement. The proactive identification of stability issues, solubility concerns, and compatibility issues enables formulation adjustments that enhance drug performance. Ultimately, the thorough understanding provided by pre-formulation studies contributes to the development of stable, bioavailable, and effective drug formulations, fostering success in subsequent stages of drug development and ensuring better clinical outcomes
REFERENCE






Adithi P*, Aarthi S, Swetha P, Aimen Bashir, Vignesh C, Sujan Kumar P, A Comprehensive Review On Pre-Formulation Strategies For Dosage Forms, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 8, 2658-2666. https://doi.org/10.5281/zenodo.13236071
 10.5281/zenodo.13236071
10.5281/zenodo.13236071
