1,3 Health Science Center — School of Pharmacy, Xi'an Jiaotong University
2 School of Foreign Languages and International Relations, Henan, China
4,5,6,7,8,9,10 School of Pharmaceutical Science, Zhengzhou University, Henan, China
11 Artificial Intelligence, Shanghai University of Engineering Science
Ferroptosis is a unique type of controlled cell death that is dependent on iron and is distinguished by the buildup of lipid peroxides. Its distinct morphological, biochemical, and genetic traits set it apart from other types of cell death, including necrosis, autophagy, and apoptosis. Recent research has shown its crucial role in the development, progression, and emergence of treatment resistance in a number of cancers, most notably Gastric cancer (GC), a highly morbid and fatal disease that is common around the world. With an emphasis on iron metabolism, lipid peroxidation, and the critical regulatory functions of glutathione peroxidase 4 (GPX4), solute carrier family 7 member 11 (SLC7A11), and acyl-CoA synthetase long-chain family member 4 (ACSL4), this review provides a thorough investigation of the molecular mechanisms underlying ferroptosis. It also looks at how oncogenes, such as p53, KRAS, and MYC, and tumor suppressors affect ferroptosis sensitivity. The review also addresses ferroptosis’s important therapeutic implications in GC and emphasizes its emerging significance in overcoming chemoresistance. The ability to promote ferroptosis and enhance the effectiveness of well-established chemotherapy and immunotherapy procedures has been shown for both natural substances, such as curcumin and artesunate, and synthetic drugs, such as erastin and RSL3. The essential functions of non-coding RNAs, particularly circular RNAs (circRNAs), long non-coding (lncRNAs), and microRNAs, in modifying ferroptosis pathways are also clarified, indicating their potential as useful biomarkers and prospective therapeutic targets. Despite these significant advancements, systemic toxicity, lack of selectivity, and the complex effect of the tumor microenvironment remain obstacles to the clinical translation of ferroptosis-based treatments. Thus, this analysis highlights the urgent need for more thorough research into new ferroptosis-based treatments and the creation of reliable prognostic biomarkers. In the end, Ferroptosis targeting is a very promising approach to improving the results of GC treatment and making it easier to apply individualized therapeutic measures.
INTRODUCTION
Cell Death a Fundamental Biological Process
Cell death, which eliminates damaged or unnecessary cells, is essential for forming tissues and organs during development and preserving tissue balance after birth. Additionally, it prevents the spread of infections by eradicating contaminated cells [1]. Both normal physiological functions and disease processes are aided by cell death. The life of a cell can range from a few days to many years, with significant variation based on the type. Programmed cell death is necessary for many vital physiological functions, including embryonic development and immune system B and T cell selection [2]. The idea of cell death was not widely accepted by biologists and researchers until the 19th century, when death was only understood at the level of individual organisms [3]. Cellular Death was identified with the development of light microscopy, tissue sectioning procedures, and staining methods [4]. The journal Cell Death & Differentiation published “Molecular mechanisms of Cell Death: recommendations of the Nomenclature Committee on cell death 2018” in 2018 as the outcome of a cooperative effort involving hundreds of cell death researchers. Cell death was divided into two main categories: Accidental Cell Death (ACD) and Regulated Cell Death (RCD) [5, 6]. An unplanned cellular death brought on by unexpected, harmful events is known as Accidental Cell Death. RCD, on the other hand, is distinguished by carefully coordinated signaling pathways that are critical for tissue regeneration and the developmental process [7, 8]. Regulated cell death mediated by dedicated molecular mechanisms, known as programmed cell death, plays important roles in health and disease maintenance [9]. It also relies on specific molecular machinery and is not the same as classical ACD Necrosis, which is defined as uncontrolled cell death brought on by severe biological, chemical, or physical insults [10]. One of the established mechanisms of regulated cell death (RCD) is Apoptosis [11, 12]. RCD forms are categorized based on their morphotypes into three categories: (i) Apoptosis or type I cell death, (ii) Autophagy or type II cell death, and (iii) Necrosis or type III cell death [13]. Apoptosis is a metabolically active type of cell death that is distinguished by specific morphological alterations, such as nuclear fragmentation, chromatin condensation, cell shrinkage, and blebbing of the plasma membrane [14]. Autophagy is a catabolic process that occurs in cells. Autophagy is classified as a form of RCD due to its frequent co-occurrence; autophagy can also influence the regulation of inflammation [15]. Beyond apoptosis, numerous other forms of RCD have been identified, including Autophagic Cell Death[16], Lysosomal Cell Death [17], Mitoptosis [18], Paraptosis [19], Pyroptosis [20], NETosis [21], Necroptosis [22], Immunogenic Cell Death [23], Entosis [24], mitochondrial permeability transition (MPT)-dependent necrosis [25], Methuosis [26], Parthanatos [27], Ferroptosis [28], Autosis [29], Alkaliptosis [30], Oxeiptosis [31], Cuproptosis [32], and Erebosis [33] (Figure 1).
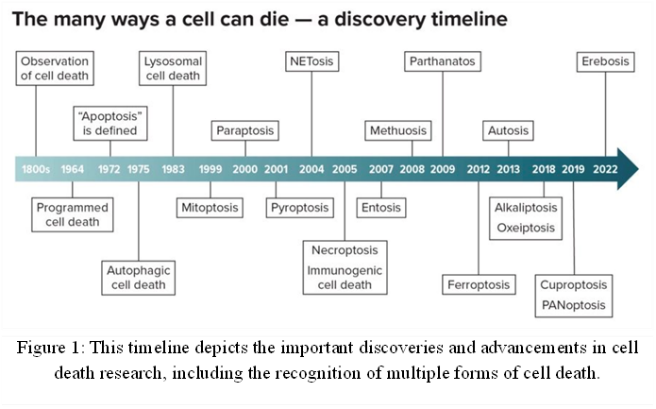
However, disruptions in cell death signaling can contribute to the development of various human diseases [34]. Uncontrolled, excessive, or poorly regulated cell proliferation can result in illnesses like inflammation [35], cancer [36] and even death for the organism. In contrast, diseases like Rheumatoid Arthritis, Parkinson’s disease, and Alzheimer’s disease [37] have been connected to an excessive rate of cell death [38].
Ferroptosis a Distinct Form of Regulated Cell Death
Ferroptosis, derived from the Greek word “ptosis” (falling) and the Latin word “ferrum” (iron) [39], is a newly identified form of controlled cell death that differs from other cell death mechanisms in its morphology, biochemistry, and genetic makeup [40, 41]. Dixon first proposed the idea of Ferroptosis in 2012. He described it as a mechanism of non-apoptotic cell death that is dependent on iron and characterized by the accumulation of lipid reactive oxygen species (ROS) [42]. Iron is vital for cellular functions; however, accumulating excessive iron can trigger various forms of cell death, including ferroptosis [43]. The average daily loss of iron in the human body is 1-2 mg, and the body normally maintains a total iron content of 3-4 grams [44]. Iron absorption primarily comes from dietary intake, mainly in the form of Fe3+. Within the body, haemoglobin is the largest iron storage unit [45]. Ferroptosis may have been first proposed as a theory based on observations of cancer cell from nutrient (specifically cysteine) deprivation and “Oxytosis” a type of neuronal cell death caused by the excitotoxin glutamate in conjugation with inhibition of the amino acid antiporter solute carrier family 7 member 11 (SLC7A11/xCT/system xc-) [46, 47]. This unique type of cell death is influenced by a number of cellular metabolic processes and is caused by iron-dependent phospholipid peroxidation [48]. These include many signaling pathways that are relevant to disease, as well as redox homeostasis, and iron regulation [49], mitochondrial function [50], and the metabolism of sugars [51], lipids, and amino acids [52]. Research on ferroptosis over a number of years has proven the critical role that mitochondria play in ferroptosis through their energetic metabolism, iron metabolism, lipid metabolism, and other regulatory functions [53]. Other organelles take part in ferroptosis too, besides mitochondria. Lysosomal dysfunction and oxidative stress are linked to the endoplasmic reticulum [54], and lipid peroxidation brought on by Golgi stress [55] all play a part in the initial phase of this cell death process [39]. Ferroptosis is primarily regulated by Glutathione peroxidase 4 (GPX4), a significant lipid peroxidation scavenger [30]. This protein, which is selenium-dependent, uses glutathione (GSH) to lower lipid peroxides and shield cells’ membranes [56]. System Xc- mediated cystine uptake and GSH synthesis sustain GPX4 activity, whereas ferroptosis is triggered by ferroptosis or its inhibition (e.g., by RSL3) [57, 58]. Remarkably, ferroptosis resistance is a result of both mitochondrial and cytoplasmic GPX4 isoforms, with the cytoplasmic form being crucial for embryonic development. There are two primary ways that ferroptosis can happen in cells. The first is to target either the amino acid antiporter system xc- [59] or iron-transporting molecules like lactotransferrin (LTF) [60] and Transferrin (TF) [61, 62]. The presence of redox-active iron and the ensuing iron-dependent peroxidation of polyunsaturated fatty acids within phospholipids found in cell membranes are its main characteristics [63]. Because glutathione peroxidase 4 (GPX4) is inactivated during this process, lipid-based reactive oxygen species accumulate [64].
Table 1: Hallmarks of Ferroptosis Types of Regulated Cell Death
|
Type |
Morphological features [61, 65-70] |
Biochemical features [71-80] |
Immune features [81, 82] |
Major regulators ADDIN EN.CITE.DATA [7, 53, 54, 83-92] |
Major inhibitors [31, 57, 64, 93-98] |
|
Ferroptosis |
Smaller mitochondria, reduced mitochondrial cristae, elevated mitochondrial membrane densities, increased rupture of mitochondrial membranes |
Iron Accumulation, Lipid peroxidation, ΔΨm dissipation, MAP1LC3B-I to MAP1LC3B-II conversion, glutaminolysis, caspase-independent |
ICD |
Positive: TFRC, ACSL4, LPCAT3, ALOX15, GLS2, DPP4, NCOA4, BAP1, BECN1, PEBP1, CARS, VDAC2/3, RAB7A, HSP90, and ALK4/5
Negative: SLC7A11, GPX4, NFE2L2, HSPB1, HSPA5, FANCD2, NFS1, ITGA6, ITGB4, and OTUB1
Dual: TP53 |
Deferoxamine (Fe), Cyclipirox (Fe), Deferiprone (Fe), Ferrostatin-1 (ROS), Liproxstatin-1 (ROS), β-mercaptoethanol (ROS), Vitamin E (ROS), β-carotene (ROS), NAC (ROS), XJB-5-131 (ROS), Zileuton (ROS), CoQ10 (ROS), Baicalein (ROS), Vildagliptin (DPP4), Alogliptin (DPP4), Linagliptin (DPP4), Thiazolidinedione (ACSL4), Rosiglitazone (ACSL4), Selenium (GPX4) |
During ferroptosis, the internal oxidation and reduction balance of the cell is disrupted. In particular, oxidizing agents such as ROS and divalent iron ions start to accumulate while protective antioxidants such as GSH and GPX4 start to decrease [99]. Ferroptosis is increasingly associated with ischemia-reperfusion injury [100], stroke [92], traumatic brain injury [101], neurodegenerative diseases including a rare form of Parkinson’s disease (PD) [102, 103], and the development of cancer [104]. It also has great therapeutic potential for the treatment of cancer and other illnesses [105] (Figure 2).
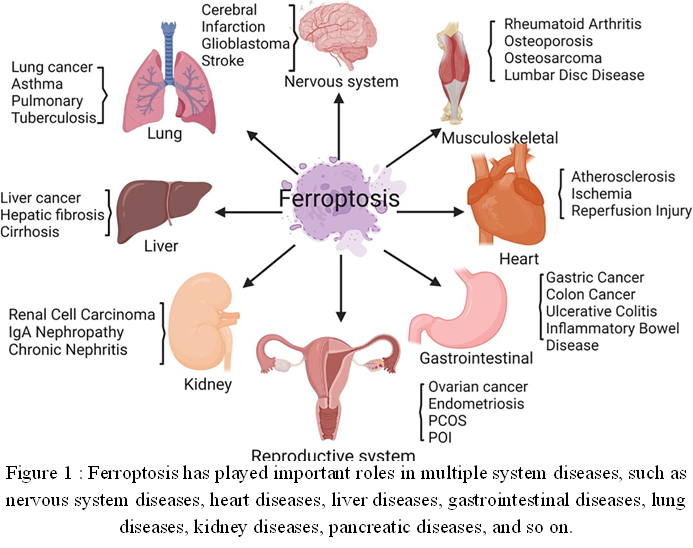
Figure 2 : Ferroptosis has played important roles in multiple system diseases, such as nervous system diseases, heart diseases, liver diseases, gastrointestinal diseases, lung diseases, kidney diseases, pancreatic diseases, and so on.
Gastric Cancer a Global Health Burden
Gastric Cancer (GC) is the fifth most common cause of cancer-related deaths and the fifth most common cancer diagnosed globally, accounting for over one million new cases and 769000 annual deaths [106]. It is a very deadly cancer. Less than 10% of GV patients survive for five years, which is generally a poor prognosis [80]. The pathogenesis of gastric cancer is complicated and involves a host of genetic, environmental, age, and microbiological factors [107, 108]. Gastric cancer is often diagnosed at a larger stage because there are no obvious early signs, which means that many patients present with an incurable disease [109]. Only 30%-40% of patients survive for five years after diagnosis due to the subtle and gradual development of gastric cancer, which makes early detection challenging [110]. In South America, Eastern Europe, and East Asia, there are higher incidence and mortality rates of Gastric Cancer [111] (Figure 3) .
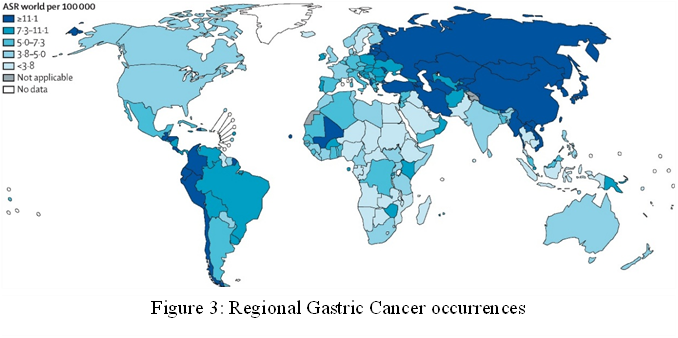
In addition, Eastern Europe has higher age-standardized incidence rates of gastric cancer than Central and Western Europe [112]. GC can be classified histologically using different classification systems, but according to the Laurén classification system, gastric cancer has historically been divided into three main histological subtypes: Diffuse-type, Intestinal-type, and mixed [113, 114]. The Japanese Gastric Cancer Association, Ming, Goseki, and Nakamura have all proposed additional classification systems for Gastric cancer. Although they acknowledge the existence of other, less noticeable components, these classifications are mainly based on the predominant histological type found in the tumor [115]. The subtypes account for about 30%, 50%, and 20% of cases, respectively. The clinicopathological features of intestinal-type (ITGC) and diffuse-type gastric cancer (DTGC) are very different [116] (Figure 4).
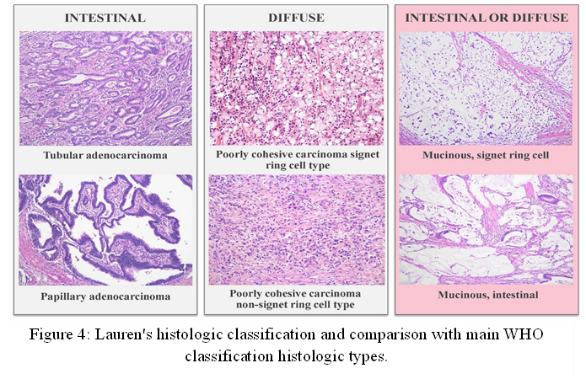
Intestinal-type gastric cancer (ITGC) is most commonly linked to chronic infection with Helicobacter pylori (HP) and represents the final stage in a neoplastic progression known as Correa’s cascade [117]. Atrophic gastritis (AG) and intestinal metaplasia (IM) are precancerous conditions that can result from chronic inflammation of the stomach lining brought on by HP colonization [118]. AG and IM are both known risk factors for ITGC [119]. A higher incidence in men, occurrence at an older age, and a propensity to develop in the lower (distal) part of the stomach, the first site of HP infection, are additional features of IGTC [120]. The intestinal subtype of gastric cancer can be further classified into different molecular subtypes. These consists of the following: (I) Cancers associated with Epstein-Barr virus (EBV); (II) Cancers that are mismatch-repair-deficient (MMR-D); (III) Cancers that have TP53 aberrations; and (IV) a category that lacks any of these particular characteristics and is frequently referred to as “other” [121]. Diffuse-type gastric cancer, also called scirrhous gastric cancer, is distinguished by a tissue composition where cancer-associated fibroblasts (CAFs) are more abundant than the actual cancer cells [122]. In the early stage of diffuse-type gastric cancer (DGC), mutated cells separate from the stomach lining and form tiny lesions in the mucosa, the innermost layer. Signet ring cells (SRCs) are found sporadically in these mucosal lesions. Mucin, which fills the cytoplasm of SRCs, pushes the nucleus to the side, giving it a crescent shape [123]. Furthermore, the Asian Cancer Research Group (ACRG) has identified the microsatellite stable and epithelial-to-mesenchymal transition (MSS/EMT) subtype as the one linked to the worst patient outcomes among diffuse-type gastric cancers (DGCs) [124]. Besides, Diffuse-type gastric cancer tissue comprises seven main cell types: epithelial cells, fibroblasts, endothelial cells, and various immune cells, including T cells, B cells, Plasma cells, and myeloid cells [125].
Table 2: Key differences in clinicopathologic characteristics of diffuse-type and intestinal-type gastric cancer
|
Features |
Diffuse-type gastric cancer |
Intestinal-type gastric cancer |
References |
|
Prevalence |
~30% |
~50% |
[99, 126] |
|
Age |
Younger |
Older |
[126, 127] |
|
Sex |
Female incidence = Male incidence |
Male predominant |
[48, 128] |
|
Genetic risk factors |
Strong |
Weak |
[129] |
|
Environmental risk factors (Helicobacter pylori infection, smoking, alcohol, and obesity) |
Modest |
Strong |
[81, 130, 131] |
|
Pathology characteristics |
Poorly cohesive, signet ring cells |
Tubular papillary |
[123] |
|
Type of metastasis |
Peritoneal |
Liver |
[132] |
|
Prognosis |
Worse |
Better |
[124] |
Currently, the main approach to treating gastric cancer (GC) is endoscopy for detection, followed by a gastrectomy (The surgical removal of the stomach) and chemotherapy (CT) or chemo-radiotherapy (CRT) either before or after the procedure [133]. Although there have been notable developments in CT, CRT, and immunotherapy, the only potentially effective treatment for GC is now surgical tumor removal [134]. With improvements in surgical methods, conventional chemotherapy and radiation, and the use of neoadjuvant therapy, the 5-year survival rate for gastric cancer in its early stages can surpass 95% [135]. Because gastric cancer often spreads to the lymph nodes, dissecting the lymph nodes has become an essential part of surgical treatment in GC [134]. Moreover, the surgical treatment of GC is gradually moving into the era of precision medicine due to the development of modern imaging technology, as well as the rise of artificial intelligence and big data [136]. As our understanding of Gastrin Cancer at the molecular level deepens, we are realizing that these cancers are part of a broad category of illness, each with a remarkably unique underlying mechanism of development and active pathways that promote cancer [137].
Scope of the Review
Ferroptosis is a promising but little-studied approach to studying Gastric Cancer (GC), filling in knowledge gaps about lipid peroxidation, iron metabolism, and iron imbalance in carcinogenesis. Different from apoptosis, necrosis, and autophagy, its unique mechanistic profile presents new therapeutic opportunities, especially for aggressive subtypes like diffuse-type GC that do not yet have targeted strategies. Important questions remain unanswered despite the growing recognition of Ferroptosis’s role in GC's pathophysiology, progression, and treatment resistance. In this review, we studied that ferroptosis may be a significant factor in the development of gastric cancer, treatment resistance, and overall patient outcomes. Thus, clarifying the mechanisms and consequences of Ferroptosis in relation to gastric cancer may provide new information for prognosis, diagnosis, and treatment. The objective of this review is to thoroughly examine the state of knowledge regarding Ferroptosis, its molecular regulators, and its possible involvement in the etiology and therapeutic targeting of gastric cancer.
The Molecular Mechanisms Underlying Ferroptosis
Ferroptosis, a non-apoptotic form of controlled cell death marked by iron-dependent lipid peroxidation, has been linked to a number of metabolic diseases and disturbances in the equilibrium of cells [30]. Ferroptosis is a unique type of cell death that can be brought on by administering certain small molecules, like RAS-selective lethal 3 (RSL3) and erastin [138]. The capacity of these molecules to target tumor cells in mammals with an overactive mutant RAS oncogene led to their particular selection [139]. Briefly, when too much Hydrogen Peroxide (H2O2) reacts with iron in an iron-rich environment, it produces extremely reactive hydroxyl radicals through the Fenton reaction. In order to cause membrane breakdown and ferroptotic cell death, these radicals then target and peroxidize the bis-allylic sites of phospholipids in the cell membrane that contain polyunsaturated fatty acids (PUFAs) [60]. Here we provide a brief overview of the mechanisms and key regulators of ferroptosis, emphasizing the functions of iron, amino acid metabolism, and reactive oxygen species, particularly lipid peroxides. Additionally, we highlight the possibility of using Ferroptosis as a cancer treatment tactic [140].
Mechanisms of iron accumulation during Ferroptosis
Cell growth is supported by physiological iron levels, but alterations in its redox state (Fe2+ or Fe3+) can alter a cell’s vulnerability to cell death, including Ferroptosis [141]. About 25% of the body’s iron is stored as ferritin and hemosiderin in the liver, spleen, and bone marrow, while the remaining 60% is bound to erythrocytes. Less than 0.1% of the iron in the body is carried by transferrin in the blood, while the remaining 5% is found in myoglobin [142]. When hydrogen peroxide and Fe2+ combine, the result is the Fenton reaction, which generates hydroxyl radicals and extremely reactive hydroxides [143]. Methods that raise intracellular free iron levels can accelerate the Fenton reaction, which produces ROS and encourages Ferroptosis [144]. Fenton reaction caused by ferrous (Fe2+) causes ferroptosis to begin, which results in the buildup of Reactive oxygen species (ROS) [145]. The process of iron metabolism [72]- absorption, storage, utilization, and export are strictly regulated [146]. Reduction to Fe2+ is necessary for the absorption of dietary nonheme iron, which is primarily in the insoluble Fe3+ form [147]. When in circulation, Fe3+ binds to transferrin (TF), which the cell membranes’ transferrin receptor (TFRC) recognizes [98]. Iron absorption and the subsequent generation of lipid ROS are further regulated by the phosphorylation-dependent cytoskeletal regulation mediated by heat shock protein family B (small) member 1 (HSPB1, or HSP27 in humans) [69, 148]. Therefore, Ferroptosis sensitivity is decreased by overexpressing HSF1-dependent HSPB1 or genetically suppressing TFRC [149, 150]. On the other hand, lactotransferrin (LTF) might promote Ferroptosis and iron absorption without the aid of TFRC [151] (Figure 5).
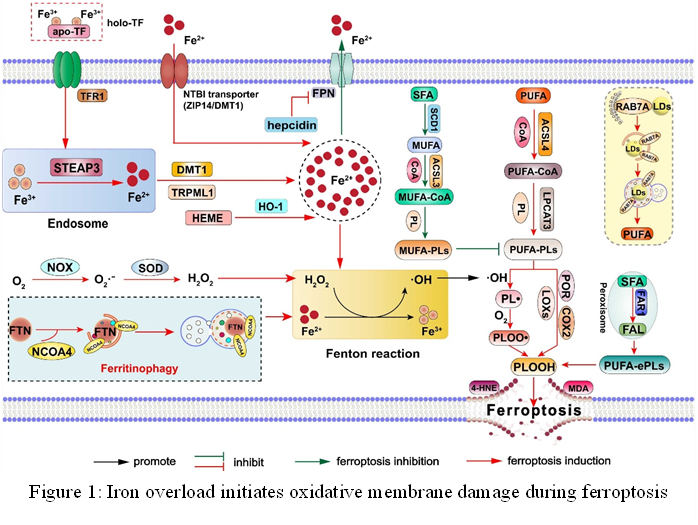
Figure 5: Iron overload initiates oxidative membrane damage during ferroptosis
Following iron absorption, Ferroptosis is believed to be facilitated by increased intracellular free iron levels due to STEAP3-mediated endosome iron reduction and DMT1 (SLC11A2)-dependent Fe2+ release into the cytosol [152]. Within the cytoplasm, the maximum number of Fe2+ is sequestered within ferritin, with a smaller quantity found in the labile iron pool (LIP), which is a form that is easily accessible [153]. Ferroptosis is also facilitated by limiting iron use for iron-sulfur cluster synthesis and lowering iron shortage through ferritinophagy-mediated ferritin degradation [100, 154]. Ferritin is a complex of 24 subunits, mostly of two types: the light subunit (FTL) and the heavy subunit (FTH1) [155]. Ferritin can be broken down by autophagy. Nuclear receptor coactivator 4 (NCOA4), a specific ferritin receptor, mediates the autophagic breakdown [156]. Similarly, it has been demonstrated that in some circumstances, increasing Ferroptosis susceptibility can be achieved by blocking PROM2- and LCN2-mediated iron efflux or by inhibiting iron export through autophagic degradation of ferroportin-1 (SLC40A1) [64, 157]. Further favoring Ferroptosis is the inhibition of mitochondrial iron exporters (CISD1/mitoNEET or CISD2), which increases mitochondrial iron retention [153, 158]. All of these results point to a very intricate network of iron metabolism that regulates cellular iron levels, which in turn affects the Fenton reaction and subsequent generation of ROS during Ferroptosis. Notably, iron is also necessary for a large number of metabolic enzymes that are redox active and involved in lipid peroxidation [159]. It is still difficult to determine whether iron’s function in controlling Ferroptosis results from the production of ROS or from iron-dependent enzyme activity. Although other metals, such as copper and zinc, can also generate reactive oxygen species (ROS) through Fenton-like reactions [160], preliminary research revealed that Ferroptosis is only induced by iron, not by other metals. This raises concerns regarding the Fenton reaction’s distinct role in this process [92]. Moreover, it is still unknown which particular chemicals or signals determine whether cells react to iron by surviving or dying.
Lipid Peroxidation in Ferroptosis: Mechanisms and Regulation
Ferroptosis is characterized by three key features: the oxidation of phospholipids containing polyunsaturated fatty acids (PUFA), the presence of redox-active iron, and the failure of lipid peroxide scavenging [110]. Lipid peroxidation, which primarily targets polyunsaturated fatty acids (PUFAs), especially arachidonic acid (AA) and adrenic acid, is a characteristic of Ferroptosis [161]. Polyunsaturated fatty acids are linear fatty acids with 18-22 carbon atoms and two or more double bonds. Usually, they are divided into two families: omega-3 [162] and omega-6 [4]. Alpha-linolenic acid [163], eicosapentaenoic acid (EPA) [164], and docosahexaenoic acid (DHA) [165] are important members of the omega-3 family. The two main components of the omega-6 are arachidonic acid (AA) [166] and linoleic acid (LA) [167]. Because of their high reactivity, the hydrogen atoms on the methylene group (-CH2-) that sits between two double bonds in PUFA molecules are more prone to peroxidation [168]. Three phases can be used to describe the free radical reactions involved in lipid peroxidation (LPO) of biological membranes: (I) initiation, in which a phospholipid containing a polyunsaturated fatty acid (PL) has a hydrogen atom removed by an initiating free radical, forming phospholipid carbon radical (PL*), which subsequently reacts with oxygen to form a hydroperoxyl radical (PL-OO*) [169]; (II) chain propagation, in which the PL-OO* reacts with another PL to produce phospholipid hydroperoxide (PL-OOH); and (III) arrest, which takes place as a result of free radical scavenging or radical-radical interactions [170] (Figure 6).
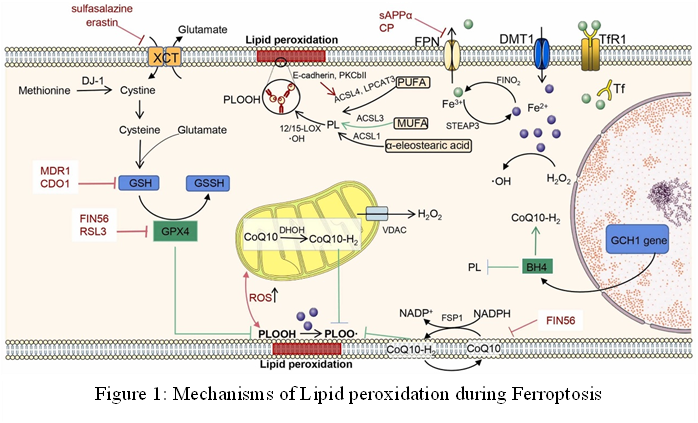
Figure 6: Mechanisms of Lipid peroxidation during Ferroptosis
This process starts when acyl-CoA synthetase long-chain family member (ACSL4) esterifies PUFAs with CoA to create PUFA-CoA [22]. Following this, Lysophosphatidylcholine acyltransferase 3 (LPCAT3) causes PUFA-CoA to be incorporated into membrane phospholipids (PLs), resulting in PUFA-containing phospholipids (PUFA-PLs) [5]. The PUFA-PLs are subjected to enzymatic peroxidation by cytochrome P450 oxidoreductase (POR) and lipoxygenases (LOXs), which results in the production of reactive oxygen species (ROS) and phospholipid hydroperoxides (PL-PUFA-OOH) [171, 172]. In parallel, the Fenton reaction, which is fueled by Fe2+ and superoxide anions, can cause the non-enzymatic peroxidation of PUFA-PLs [173]. Lipid peroxide buildup eventually weakens membrane integrity and causes plasma membrane rupture, which is a hallmark of ferroptotic cell death [174]. One effective method for inhibiting ferroptosis is to target LPCAT3 and ACSL4 [100, 175]. By changing the intracellular PUFA-phospholipid composition, which is marked by a decrease in C20:4 species and an increase in C22:4 phospholipids, inhibition of LPCAT3 increases the resistance of LPCAT3-deficient cells to ferroptosis [176, 177]. Additionally, lipid peroxidation-PKCβII-ACSL4 creates a positive feedback loop that may be a therapeutic target because protein kinase CβII (PKCβII) amplifies peroxidation by activating ACSL4 and acts as a sensor of early lipid peroxidation events [7, 178]. On the other hand, by competing with PUFAs for inclusion in phospholipids, monounsaturated fatty acids (MUFAs) act as organic ferroptosis inhibitors [179]. In addition to SCD/SCD1-driven CoQ10 production, MUFA-mediated protection includes the reduction of ROS and PUFA-PL accumulation at the membrane, which is dependent on ACSL3 [180, 181]. Furthermore, MUFAs strengthen membrane integrity by reducing unsaturated acyl chains in phospholipids and encouraging the synthesis of long-chain saturated ceramides [182]. Ferroptosis resistance has been linked to both endogenous MUFA production and dietary MUFA intake, underscoring their potential as therapeutics [183]. Although Ferroptosis ultimately results in cell death due to lipid peroxidation, the exact series of events that trigger this process is still being studied [184]. Lipid ROS can accumulate as a result of the oxidation of PUFAs in membrane phospholipids caused by an excess of reactive oxygen species brought on by an imbalance in the cellular redox system [185, 186]. The Fenton reaction, which is catalyzed by Fe2+, can speed up this oxidative process even more [187]. However, Glutathione peroxidase 4 is essential for preventing the production of these harmful lipid peroxides [188]. Lipid metabolism [189], iron metabolism [190], and intracellular redox homeostasis [32] thus interact intricately in lipid peroxidation, which is known as the final event in Ferroptosis . Furthermore, it has been demonstrated that changes in the amounts of PUFA in cell membranes affect a cell’s vulnerability to Ferroptosis [191].
Ferroptosis in Cancer
Ferroptosis has a complicated and dual function in cancer, serving as a target for resistance mechanisms as well as a possible vulnerability [192]. Due to oncogenic signaling pathways that mostly rely on redox-dependent signaling, cancer cells frequently display high rates of proliferation and redox imbalance [193]. Furthermore, tumors’ iron-rich microenvironment encourages oxidative stress and fast cell proliferation, which makes them dependent on mechanisms that prevent ferroptotic death [108]. This resistance to Ferroptosis promotes chemoresistance and offers a growth benefit [113]. For example, glutathione and thioredoxin systems are often elevated to combat oxidative damage, and somatic mutations in Nrf2 [194]or keap1 [195] that are frequently detected in malignancies, and expression of antioxidant enzymes [196]. The fact that cancer cells with oncogenic RAS mutations are especially vulnerable to Ferroptosis inducers like erastin and RSL3 led to the identification of Ferroptosis itself [197, 198]. This sensitivity is associated with activation of the RAS-RAF-MEK pathway [199], which enhances iron intake by suppressing ferritin and upregulating transferrin receptors [186]. Nonetheless, Ferroptosis sensitivity differs between cancer types and can depend on the situation [61, 151, 200]. Recent research further links the regulation of Ferroptosis to tumor suppressors like p53 [201]. p53 can promote Ferroptosis by downregulating SLC7A11 [202], a critical component of system Xc-, hence restricting cystine absorption and increasing sensitivity to lipid peroxidation [100]. However, p53 has the ability to boost antioxidant defenses [203], which enables cancer cells to avoid Ferroptosis [59, 204]. Additionally, GPX4 and system Xc- are highly dependent in some malignancies [205], including triple-negative breast cancer and B-cell-derived lymphomas, indicating that these pathways could be therapeutic targets [43, 206]. Ferroptosis modulation is a viable cancer treatment option because of the dual nature of Ferroptosis in cancer, which reveals both a possible tumor suppression mechanism and an adaptive strategy of cancer cells to withstand oxidative death [207, 208].
Gastric Cancer: Basics and Causes
Genetics of Gastric Cancer
About 1-3% of patients with gastric cancer have a germline mutation [209], and 10% of all gastric cancers show familial aggregation [210]. There are three types of hereditary gastric cancer [211]: familial intestinal gastric cancer (autosomal dominant) [212], adenocarcinoma with proximal polyposis of the stomach (autosomal dominant) [213], and hereditary diffuse-type gastric cancer (HDGC; autosomal dominant; less than 1% of all gastric cancer causes) [214]. The CDH1 gene, which codes for E-cadherin, has mutations in 30-40% of HDGC patients [215]. Individuals with harmful CDH1 mutations should be treated at specialist centers, and those who fit the screening criteria for CDH1 genetic testing should be sent to a clinical geneticist [216]. People with harmful CDH1 mutations should have a prophylactic complete gastrectomy [217]. A germline mutation in CTNNA1, the gene encoding αE-catenin, has also been found in three HDGC families [218]. The insulin receptor gene, PALB2, FBXO24 (F-box protein 24), and DOT1L (DOT1-like histone H3K7 methyltransferase) are further putative candidate genes linked to HDGC [219, 220]. However, the underlying genetic change in about 70% of HDGC patients is still unclear [221]. Certain point mutations in the APC promoter 1B gene have been linked to Gastric Cancer with proximal polyposis of the stomach [222]. Furthermore, people with other heredity conditions like lynch syndrome (linked to mutations in MLH1, MSH2, PMS2, and MSH6) [223], Cowden syndrome (PTEN) [224], juvenile polyposis (BMPR1A and SMAD4) [225], Li-Fraumeni syndrome (TP53) [226], Peutz-Jeghers syndrome (STK11) [227], familial adenomatous polyposis (Caused by APC mutations), and familial adenomatous polyposis are more likely to develop gastric cancer [228].
Helicobacter Pylori
It has been determined that the most common pathogenic factor causing Gastric cancer is H. pylori infection [229]. It has been officially designated as a class I carcinogen and is thought to impact a significant percentage of the world's population-more than half [230]. A small percentage of those infected-roughly 2%-follow a predictable, step pattern of illness progression, which is crucially reversible if detected early [231]. With special attention to the CagA protein, the cag pathogenicity islands are a major mechanism by which H. pylori causes cancer [81]. With the cytotoxin-associated gene A (CagA), the H. pylori cag pathogenicity island (cagPAI)-encoded type IV secretion system (T4SS) increases the risk for developing malignancies [232]. CagA, which is expressed in 70% of US and 100% of Asian H. pylori patients [233], enters host cells through the type IV secretion system and [234], depending on its post-translational status, activates signaling pathways such as SHP2, Abl, or Src kinases [10, 235]. H. pylori strains differ in their EPIYA motif, the phosphorylation site of CagA [236]. The EPIYA-ABD genotype, which is common in East-Asia, is associated with an increased risk of gastric cancer and may account for the high rates in Thailand [237]. In addition to CagA, H. pylori secretes the Vaca toxin, which inhibits T-cell responses and promotes lesion formation, and peptidoglycan, which promotes inflammation by upregulating cytokines such as IL-8 and COX [238, 239]. Notably, it has been demonstrated that treating gastric ulcer patients with a H. pylori infection reduces their risk of getting gastric cancer, with a markedly lower incidence in those who receive treatment [240].
Epstein-Barr Virus
Roughly 10% of all occurrences of Gastric cancer have been linked to the Epstein-Barr virus [241]. Although the exact mechanism of its infection has not yet been fully determined, several theories are presently being considered [242]. One important finding is that cells with high expression of the CD21 marker are more likely to be infected by EBV [119, 243]. Furthermore, the virus may enter cells by endocytosis [244], especially if it is encased in IgA antibodies that subsequently interact with cell surface IgA receptors [245]. Another theory focuses on the virus’s variety of glycoproteins, such as gH/gL and gp42, which have the ability to bind with B-lymphocytes’ HLA class II molecules [246]. Notably, the gH, gL, and gB complexes have the capacity to fuse with epithelial membranes, allowing infection, where gp42’s attachment to B-lymphocytes may prevent EBV infection in epithelial cells [247, 248]. Furthermore, it has been proposed that the crucial processes of viral fusion and subsequent cell entry may possibly be influenced by interactions between the host’s β2 integrin protein and the EBV BMFR2 protein [249].
Non-Pathogenic Influences
Apart from infectious agents, a range of non-pathogenic variables, including genetics and environmental exposures, contribute to the development of Gastric cancer [250]. About 10% of all occurrences of Gastric cancer are hereditary [251], and 3% of these cases are caused by germline mutations in the CDH1 gene, a disorder called hereditary diffuse Gastric cancer (HDGC) [252]. Prophylactic complete gastrectomy is the main preventive measure since the absence of a functional CDH1 protein causes a significantly high incidence of the disease in families with this genetic mutation [253]. Lifestyle and environmental variables can add to the total risk. These include unhealthy eating habits that involve bad decisions, smoking and excessive salt intake [184]. Notably, there is a statistically significant correlation between consuming red and processed meats and an increased risk of Gastric cancer [254]. The practice of eating late into the night, consuming foods that are too hot, and irregular sleep habits that cause changes in circadian rhythm genes are other possible contributing causes [255]. Although the majority of individuals engaged in these habits do not get cancer, certain individuals may benefit from adopting changes to their lifestyle to minimize their vulnerability [256]. Furthermore, smoking has been shown to be a risk factor for gastric cancer, especially the diffuse and cardia subtypes, with the degree of risk being directly correlated with the length of time and intensity of cigarette smoking [257].
Ferroptosis and Gastric Cancer
Recent research has demonstrated that activating Ferroptosis in cancer cells has two benefits: it can effectively stop tumor growth while also increasing the effectiveness of immunotherapy, chemotherapy, and targeted therapy [258]. Because ATF3 inhibits the Nrf2/Keap1/XcT signaling pathway and induces Ferroptosis, it can increase GC cells’ sensitivity to the chemotherapeutic medication cisplatin. Meanwhile, by secreting a variety of bioactive chemicals, including exosomes, cancer-associated fibroblasts (CAFs) encourage tumor growth and medication resistance [259]. Reports reveal that cancer cells demonstrating therapeutic resistance have shown an increased vulnerability to Ferroptosis, perhaps related to their accelerated lipid metabolism controlled by the TGF-ZEB1 pathway [260].
Non-coding RNAs associated with Ferroptosis in Gastric Cancer cells
MicroRNA
Numerous studies have proved that microRNAs, which are tiny single-stranded RNA molecules with 22-24 nucleotides, cause gastric cancer cells to undergo Ferroptosis [147]. For instance, studies have shown that miR-375 targets SLC7A11 to cause Ferroptosis in GC cells both in vivo and in vitro. A possible target for triggering Ferroptosis and reducing the stem-like properties of GC cells is the mir-375/SLC7A11 regulatory pathway [261]. Furthermore, it has been found that mir-489-3p also targets the transmembrane protein SLC7A11, confirming the mir-489-3p/SLC7A11 axis as an additional mechanism to induce Ferroptosis in GC cells [262]. According to other studies, GC cells undergo Ferroptosis when mir-221-3p is lost or downregulated because this causes activating transcription factor 3 (ATF3) to be upregulated [263]. The transcription of GPX4 and HRD1 is inhibited by increased ATF3 expression, which further limits the proliferation of GC cells [264]. HRD1 then mediates the ubiquitination and degradation Ferroptosis [265]. Moreover, it has been discovered that GC cells have high expression of mir-103a-3p, which stimulates cell division [266]. When downregulated, its target glutaminase 2 (GLS2), can promote Ferroptosis and prevent GC cell growth [267]. ATF3 has been found to be a downstream mRNA target that mir-221-3p regulates within the mir-221-3p/ATF3 pathway [264]. miRNAs have been shown in studies to control mRNA translation. They may also interact with other non-coding RNAs to cooperatively modulate Ferroptosis in GC cells [268].
Long non-coding RNAs (lncRNAs)
Ferroptosis and lncRNAs are important factors in the onset and spread of GC [201]. The roles and mechanisms of Ferroptosis-related lncRNAs in GC are highlighted in this section. Through VDAC3 ubiquitination, the lncRNA BDNF-AS drives peritoneal metastasis of GC and controls Ferroptosis through the WDR5/FBXW7 axis [51]. Experimental research has demonstrated that lncRNA LASTR modulates Ferroptosis to affect GC cell motility and proliferation [269]. Poor clinical outcomes and the onset of GC are thought to be mostly caused by GC stem cells. Exosomal lncRNA FERO, produced from GC cells, has been demonstrated to limit the tumorigenicity of GCSCs by suppressing Ferroptosis, suggesting that a targeted strategy using exosomal lncFERO/hnRNPA1/SCD1 combined with chemotherapy may serve as a promising treatment for GCSC-driven GC [270]. Furthermore, it has been discovered that lncRNAs, including A2M-AS1 [271], C2orf27A, and ZNF667-AS1, target Ferroptosis-related genes and hinder CD4+ T cell activation in GC, potentially opening up a new treatment option for GC immunotherapy [272]. To improve the prediction of overall survival in GC patients, a predictive model based on 17 Ferroptosis-related lncRNAs has been developed [273]. These lncRNAs may also have a major impact on immune infiltration in GC, which could help with treatment plans and individualized prognoses [125]. To forecast the prognosis of GC, another model based on lncRNAs linked to Ferroptosis has been created. Ferroptosis-related lncRNAs have also been connected by other prediction models to immune response, treatment resistance, and changes in the tumor microenvironment in GC [48]. Differential expression of Ferroptosis-related lncRNAs is shown at different phases of GC, suggesting that these lncRNAs may be useful in clinical settings for GC diagnosis, prognosis, and treatment [274]. All of these studies point to the potential use of Ferroptosis-related lncRNAs as useful biomarkers for drug resistance, prognosis, individualized treatment, and GC progression.
Circular RNAs (circRNAs)
Numerous circular RNAs (circRNAs) have been found to be important regulators of Gastric cancer initiation and progression [275]. Ferroptosis-related circRNAs are developing prognostic biomarkers with great potential for a variety of uses, including GC diagnosis, targeted treatment plans, tumor microenvironment comprehension, drug resistance, and immunology enhancement [276]. The functions of circRNAs linked to Ferroptosis in GC are highlighted in this section. In GC tissues and cells, circRNA circ0008035 expression in noticeably elevated [277]. By acting as a sponge for mir-599, it has been demonstrated to increase EIF4A1 expression [278], which in turn inhibits Ferroptosis and modifies the proliferation and apoptosis of GC cells [276]. On the other hand, GC tissues and cell lines have downregulated levels of circRNA circ0000190 [144], which is linked to a worse prognosis for GC patients [279]. It has been shown that circ0000190 overexpression inhibits GC cell migration and proliferation by encouraging Ferroptosis [280].
Ferroptosis and H. pylori infection
H. pylori is a grade 1 carcinogen according to the World Health Organization (WHO) [281]. Although there are many different ways that iron metabolism, H. pylori infection, and cancer formation are related, it is now thought that abnormalities in iron metabolism may cause Ferroptosis and raise the risk of GC [282]. There is still disagreement over whether H. pylori is the main cause of the aberrant iron metabolism linked to GC [283]. A greater sTfR/log ferritin ratio, increased plasma levels of soluble transferrin receptor (sTfR) [284], and iron deficiency are all linked to H. pylori infection [285], which is known to increase carcinogenesis by boosting pathogen virulence [286]. Iron deficiency promotes the release of the VacA toxin and increases the expression of the H. pylori cag pathogenicity island, both of which aid in cellular transformation [287]. H. pylori relies on transferrin for iron in order to survive because ferritin receptors are mislocalized to the basolateral membrane instead of the apical surface [288]. Furthermore, by encouraging the development and activation of TH17 cells, the interaction between dyslipidemia and H. pylori infection may aid in the advancement of GC [289, 290]. Even though it is commonly known that H. pylori, Gastric cancer, and Ferroptosis are related, more investigation is required to elucidate the underlying molecular mechanisms [291].
The Clinical Evaluation of Ferroptosis in the Detection of GC
Growth hormone levels, ROS, iron ions (specifically ferrous ions), glutathione (GSH) concentrations, and other relevant variables are measured to assess cellular activity, iron metabolism, and GSH metabolism [292]. Ferroptosis-associated indicators were objectively recorded at both staging and disease progression in order to better comprehend the pertinent molecular indicators and evaluate tumor-related markers in Gastric cancer [293]. Low-dose erastin has been shown to have cytotoxic effects on gastric cancer cells, which include the formation of ROS, decreased cellular activity, and decreased ATP generation, all of which ultimately end in cell death [294]. Immunohistochemical examination of Gastric cancer tissues has revealed a substantial overexpression of perilipin2, also referred to as adipose-differentiation-associated protein [68]. Assessing the expression levels of these linked proteins serves as a basis for determining possible therapeutic targets and regulatory genes in gastric cancer, providing important information and guidance for upcoming drug development efforts.
Drug Resistance in Gastric Cancer
In clinical oncology, the increasing resistance of GC patients to chemotherapeutic drugs like paclitaxel and cisplatin poses a serious problem and is frequently linked to a poor prognosis [295]. Mutations in genes that control apoptosis and the overexpression of glutathione (GSH) levels are frequently blamed for this chemoresistance [296]. Interestingly, Ferroptosis inducers have shown promise in combating medication resistance and are worthy of more research [297]. Cancer cells have a higher tolerance to ROS than normal cells because of genetic defects and unchecked proliferation [298]. Their capacity to maintain high amounts of the antioxidant GSH, which promotes cellular development and survival, is a major factor in their resistance [299]. Research indicates that enhancing Ferroptosis in cancer cells can increase their susceptibility to chemotherapeutic drugs by interfering with the suppression of lipid-ROS mediated by exosomes generated from cancer-associated fibroblasts (CAF) [113]. The ROS-activated GCN2-elF2α-ATF4-XcT pathway, a signaling cascade linked to cisplatin resistance and mitochondrial dysfunction, is another possible therapeutic target in GC [300, 301]. Furthermore, as elevated ROS can upset the cellular redox equilibrium and encourage cell death, lowering ROS levels may be a novel therapeutic strategy [100]. Research has shown that by controlling ROS levels, the antioxidant enzyme peroxiredoxin 2 can make AGS and SNU-1 GC cells more sensitive to cisplatin treatment [302]. Targeting Ferroptosis pathways may provide an efficient method of eradicating drug-resistant tumor cells, considering the significance that persistently elevated ROS levels play in fostering drug resistance.
DISCUSSION
Ferroptosis, a type of controlled cell death characterized by its reliance on iron and the execution of lipid peroxidation, has quickly emerged as a key area of study for Gastric cancer (GC) [32]. The complex regulatory networks that control Ferroptosis have been painstakingly analyzed in this review, exposing its dualistic nature, operating as a tumor-suppressive process and, ironically, as a possible cause of treatment resistance in some circumstances [303]. As a result, Ferroptosis presents a special therapeutic option for the treatment of GC, especially when traditional therapies prove ineffective [304]. Important molecular checkpoints that determine cellular sensitivity to Ferroptosis have been found, including acyl-CoA synthetase long-chain family member 4 (ACSL4) [305], glutathione peroxidase 4 (GPX4) [306], and solute carrier family 7 member 11 (SLC7A11) [307]. Notably, resistance to Ferroptosis and poor clinical outcomes in GC patients have been repeatedly linked to the overexpression of GPX4 and SLC7A11, which aid in the detoxification of lipid peroxides and maintain redox equilibrium [308]. By carefully targeting these molecules with inhibitors like erastin (which disables system Xc-) or RSL3 (a GPX4 inhibitor), ferroptotic cell death can be successfully induced, making them attractive targets for therapeutic intervention [156]. Ferroptosis also has complex interactions with key tumor suppressor and oncogenic signaling pathways. For example, Ferroptosis is regulated by the tumor suppressor p53 in a context-dependent manner, either by suppressing it under particular metabolic conditions or by increasing it through the downregulation of SLC7A11 [309]. On the other hand, by changing iron metabolism or upregulating the antioxidant defense system, oncogenes such as KRAS and MYC can confer resistance to Ferroptosis [310]. These findings highlight how crucial it is to carefully take into account each patient’s unique genetic and molecular background when developing treatment plans based on Ferroptosis. From a medicinal perspective, several Ferroptosis inducers, including natural substances like curcumin and artesunate and small chemicals like sorafenib [311] and sulfasalazine [312], have shown effectiveness in making GC cells more sensitive to cell death [81]. Furthermore, Ferroptosis inducers may have synergistic effects with traditional treatments such as immune checkpoint inhibitors, chemotherapy, or radiation therapy, improving treatment response and overcoming chemoresistance [313]. Notably, studies have shown that drug-resistant GC cells often have a higher susceptibility to Ferroptosis because of elevated lipid metabolism and redox imbalance, offering a targeted strategy for the removal of these otherwise difficult-to-treat cells [151]. Nevertheless, it is important to recognize the substantial obstacles and constraints that still exist in spite of these encouraging lines of inquiry. The tumor microenvironment (TME), which includes elements including stromal components, immune cell infiltration, and hypoxia, can alter Ferroptosis susceptibility in a variety of intricate ways [314]. Furthermore, there are genuine concerns about off-target effects due to the systemic toxicity and lack of specificity linked to certain Ferroptosis inducers. Additionally, finding strong and trustworthy biomarkers to precisely predict Ferroptosis sensitivity is still a crucial topic that calls for more concentrated investigation. Researchers are intensively investigating the crucial function of non-coding RNAs, such as miRNAs, lncRNAs, and circRNAs, in the complex control of Ferroptosis pathways inside GC, in addition to the direct induction of Ferroptosis [315]. These compounds exhibit an impact on gene expression and Ferroptosis sensitivity, and hold potential as both novel treatment targets and important diagnostic indicators. A thorough comprehension of these regulatory networks may make it easier to create therapies that are more specialized and precisely targeted. In order to identify the precise molecular factors that determine Ferroptosis responsiveness across various GC subtypes and to create more selective inducers with advantageous pharmacokinetic characteristics, future research should be carefully planned. Several comprehensive studies into the complex connections between Ferroptosis and immunological modulation, metabolic reprogramming, and epigenetic changes will surely advance our knowledge and eventually make it possible to include Ferroptosis modulation into precision oncology approaches.
CONCLUSION
In conclusion, Ferroptosis has become a crucial and unique type of controlled cell death that has a big impact on the etiology, development, and management of Gastric Cancer (GC). Ferroptosis, which differs from more conventional cell death processes like necrosis or apoptosis due to its lipid peroxidation and iron dependence, offers fresh insights into tumor biology and new treatment options. This research has unequivocally shown that a number of molecular pathways, such as GPX4, SLC7A11, ACSL4, and p53, which not only control redox homeostasis but also interact with important oncogenic and tumor-suppressive signaling pathways, closely regulate the induction or suppression of Ferroptosis. There is strong evidence that Ferroptosis can be used therapeutically to help GC patients overcome their medication resistance. There is a compelling case for including Ferroptosis-targeted drugs in standard therapy regimens since chemo resistant Gastric cancer cells are more sensitive to Ferroptosis inducers, such as Erastin, Rsl3, or natural substances like curcumin and artesunate. This approach could lead to the creation of less harmful and more successful treatments, especially for aggressive and advanced-stage GC, for which there are currently insufficient alternatives for precision treatment. Furthermore, novel avenues for diagnosis, prognosis, and tailored treatment have been made possible by the functions of non-coding RNAs, such as miRNAs, lncRNAs, and circRNAs, in regulating Ferroptosis in GC cells. By acting as useful indicators for Ferroptosis sensitivity and disease progression prediction, these regulatory RNAs could help tailor therapy plans. Furthermore, Ferroptosis offers a therapeutic lever to alter the tumor microenvironment, potentially enhancing the effectiveness of immunotherapy, in addition to to providing a mechanistic understanding of cellular death. Even with these encouraging discoveries, it’s critical to recognize the obstacles still in place. More thorough preclinical and clinical research is required since Ferroptosis is context-dependent, the tumor microenvironment can modify it, and Ferroptosis inducers may be systemically harmful. Further, the creation of trustworthy biomarkers and patient stratification instruments is essential for determining which patients might benefit most from Ferroptosis-based treatments. In conclusion, Ferroptosis in GC is a therapeutic target as well as a vulnerability. To reach its full potential, a more thorough and in-depth comprehension of its regulatory networks and molecular mechanisms is necessary. Transforming these findings into efficient clinical treatments that eventually increase the survival and quality of life for people with stomach cancer should be the top priority of future research.
DECLARATIONS
Conflicts of interest:
The authors declared no conflicts of interest.
Author contribution:
Md. Foysal Uddin Sarker Shanto created the document's idea, design, writing, and critical evaluation. Xu Zhitao, who helped with language curation, grammatical errors, and the sequence of the proceedings. Sahar Saberi has helped with the main manuscript writing and designed the figures. Samiul Haque, Chagouri Bayan, Atefeh Bahrami, and Chamse Doha Khodri have helped through proofreading and new insights of proceeding in the flow. Hussein Mudher Ali Al Tameemi, Intisar Kalam Ispak, and Tahsin Hannan have helped with Figure design and maintenance. Md Shakib Uddin Sarker Joy has helped with the necessary software for this writing.
Credit authorship contribution statement:
Md. Foysal Uddin Sarker Shanto: the document's idea, design, writing, and critical evaluation. Xu Zhitao: language curation, grammatical errors, and the sequence of the proceedings. Sahar Saberi: wrote the main manuscript and designed the figures. Samiul Haque, Chagouri Bayan, Atefeh Bahrami, and Chamse Doha Khodri: proofreading and new insights of proceedings in the flow. Hussein Mudher Ali Al Tameemi, Intisar Kalam Ispak, and Tahsin Hannan: with Figure design and maintenance. Md Shakib Uddin Sarker Joy: The necessary software for this writing.
Ethics approval and consent to participate:
Not Applicable
Consent for publication:
Not Applicable
Availability of data and material:
All the data used in our article are available from publicly accessible sources, such as PubMed, Elsevier, Web of Science, Springer, etc.
Funding:
No funding received.
REFERENCES











Md Foysal Uddin Sarker Shanto, Xu Zhitao, Sahar Saberi, Samiul Haque, Chagouri Bayan, Atefeh Bahrami, Chamse Doha Khodri, Hussein Mudher Ali Al Tameemi, Intisar Kalam Ispak, Tahsin Hannan, Md Shakib Uddin Sarker Joy, The Role of Ferroptosis in Gastric Cancer, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 6, 5445-5478. https://doi.org/10.5281/zenodo.15761586
 10.5281/zenodo.15761586
10.5281/zenodo.15761586
