Department of Pharmaceutics, P.S.G.V.P. Mandals’s College of Pharmacy, Shahada, Maharashtra, India 425409
Cosmetics must have both a thorough toxicological evaluation and demonstrated efficacy. Prior to the present Cosmetic regulation N°1223/2009, animal testing for cosmetic products and ingredients was prohibited in 2004 and 2009, respectively, by the 7th Amendment to the European Cosmetics Directive. To assessing the safety and effectiveness of cosmetic goods and substances, a growing variety of alternatives to animal testing have been created and approved. For instance, 2D cell culture models made from human skin can be used to assess anti-inflammatory qualities or predict the likelihood of skin sensitization; 3D models that mimic human skin is used to assess the likelihood of skin irritation; and excised human skin is the gold standard for assessing dermal absorption. Regulatory requirements, genotoxicity potential, skin sensitization potential, skin and eye irritation, endocrine properties, and dermal absorption are the main topics of this manuscript's overview of the primary in vitro and ex vivo alternative models used in cosmetic product safety testing. Each model's benefits and drawbacks in terms of cosmetic product safety testing are examined, and new technologies that can overcome these drawbacks are showcased.
Cosmetics must have a thorough toxicological evaluation in addition to demonstrated efficacy. In 2004 and 2009, respectively, the European Cosmetics Directive's 7th Amendment outlawed animal testing for cosmetic products and substances. The necessary information to demonstrate safety and substantiate the claims is then specified by European Cosmetic Regulation N°1223/2009 and the particular Regulation N°655/2013. Numerous alternatives to animal testing have been created, approved, and implemented as test recommendations for the safety assessment of cosmetic items, largely due to regulatory bodies (Figure 1). With an emphasis on regulatory requirements, genotoxicity potential, skin sensitization potential, skin and eye irritation, endocrine qualities, and dermal absorption, this study examines the primary in vitro alternative models used in safety assessment of cosmetic products and chemicals. Each model's benefits and drawbacks in terms of cosmetic product safety testing are examined, and new technologies that can overcome these drawbacks are showcased
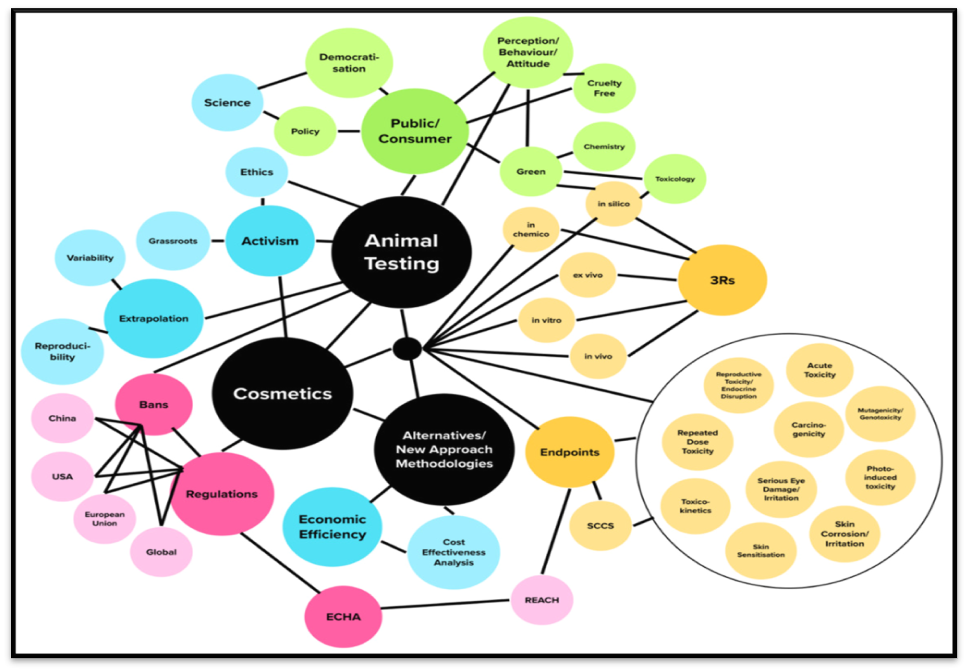
Figure 1. An overview of the various alternatives to using animals to test cosmetic items and components for safety. This review does not address assays in grey.
2. Regulatory Requirements for Cosmetics Safety Assessments
The foundation for every cosmetic product's safety in Europe is established by Cosmetic Regulation N°1223/2009 [1]. Even though many other regions lack the specific documents needed to set up their own frameworks, end consumer safety is the unifying objective of all of their policies.
Certain ingredients, such as those with particular uses (Annex VI for colorants, Annex V for preservatives, and Annex V for UV filters), must be on so-called "positive" lists.
If a component serves such a purpose, it must then meet the specifications of the specified Annex. Certain substances are restricted to specific uses (Annex III) or forbidden (Annex II).
Safety is the primary reason behind the regulation restrictions. Before being included in an annex, some ingredients in Europe are assessed by the Scientific Committee on Consumer Safety (SCCS), which then publishes its assessment along with safe usage guidelines. The SCCS releases opinions based on recommendations and the evidence that has been submitted to it. Instead of outlining the prescriptive requirement for rigorous adherence to specific regulatory "guidelines," that is beneficial. Guidelines for assessing the safety of substances are routinely provided by the European committee [2, 3]. In the USA, groups of related chemicals are typically taken into consideration based on chemical families or ingredients originating from plants by the Cosmetic Ingredient Review (CIR), which was founded by a trade association (now the PCPC) with FDA backing to prioritize and evaluate cosmetic ingredients. The risk evaluation is not included in the CIR report.

Fig2. Conducting A Cosmetic Risk Assessment
Propyl paraben as a preservative (updated opinion discarding any concern related to endocrine disruption), octocrylene as a UV filter (other update related to endocrine disruption) , and resorcinol for its use in hair dyes are just a few examples of regulated ingredients that require a positive SCCS opinion.[4]
The safety of the ingredients used in cosmetic goods may be impacted by transversal legislation. For example, the CLP Regulation (which deals with the classification, labeling, and packaging of compounds and mixtures) [5] is crucial for CMR (carcinogenic, mutagenic, and reprotoxic) substances. Carcinogenic, mutagenic, and reprotoxic substances are regarded as the most dangerous substances; in Europe, their harmonized classification is based more generally on animal experimental results (musk xylene, Disperse Yellow 3, etc.) than on epidemiological data (asbestos, benzene, etc.).[6]
Substances restricted by an Annex
When the SCCS receives a mandate from the European Commission to assess the safety of a substance for a regulated function, the opinion is based on the analysis of the scientific dossier submitted by the industry
Every endpoint is taken into account by the scientific opinion, including genotoxicity, systemic toxicity, including reprotoxicity, sub-chronic/chronic toxicity, and local tolerance (skin irritation, phototoxicity, when applicable). To determine the SED (systemic exposure dose), cutaneous absorption must be characterized.
When a wide range of applications is anticipated, as with a preservative, the exposure of the material is regarded as its expected concentration in cosmetic products, either in a single product or in several products.
As mandated by Annex I and the Guidelines [7], any other material, ingredient, or impurity must be safe for the user based on the toxicological profile, using frequently updated data from suppliers or literature. The supplier of the ingredient and the Responsible Person, who is the legal body in Europe in charge of the product and is typically the manufacturer, have two points of view about a cosmetic product that uses the ingredient. Their regulatory responsibilities are different.
Nonetheless, their goal ought to be the same: protecting consumers. Any supplier of a cometic ingredient, including businesses that produce and distribute substances within the EU, is required to register their products based on their yearly tonnage. The number of toxicological results needed in a REACH registration dossier depends on the yearly tonnage, even if a substance's intrinsic toxicity is unrelated to its production. The standards for highly hazardous and low-toxicity compounds are the same; however, substances of extremely high concern should be given special consideration within the SVHC program. Substances registered below 1 to 10 tpa (ton per annum) are exempt from the requirement for toxicological data, and as tonnage bands increase, more information must be provided. Data for in vitro skin irritation/corrosion, in vitro eye irritation, skin sensitization, in vitro bacterial gene mutation, acute toxicity, and short-term toxicity (28 days) are among the toxicological requirements for tonnages between 10 and 100 tpa (Annex VII).
Data for in vitro mutagenicity studies in mammalian cells or micronucleus studies, in vitro gene mutation studies in mammalian cells, in vitro skin irritation, in vitro eye irritation, potentially testing proposals for in vivo genotoxicity, acute toxicity, and screening for reproductive/ developmental toxicity are among the toxicological requirements at 10 to 1000 tpa (Annex VIII).
The following endpoints are included at 100 to 1000 tpa (Annex IX): extended one-generation reproductive toxicity, prenatal developmental toxicity in one species, and sub-chronic toxicity (90 days).Lastly, if activated, a long-term repeated dosage toxicity (≥ 12 months) exceeding 1000 tpa (Annex X) causes carcinogenicity, prolonged one-generation reproductive toxicity, and developmental toxicity in a second species.
We might include one of the most recent assessments of these techniques, which concentrated on REACH and cosmetics [8].
It is then crucial to understand that for compounds produced at temperatures below 10 to 1000 tpa, no information regarding DNA damage (micronucleus test) is available, and for substances produced at temperatures below 100 tpa, neither sub-chronic toxicity nor information regarding the complete reproduction cycle are known. The necessity of cosmetic companies (or responsible persons in general) to demonstrate the safety of each ingredient should then be considered by a provider of cosmetic ingredients. The product is the responsibility of the cosmetic brand (the Responsible Person). Research can be conducted on the product to verify its high level of human acceptability. [9]
Such tests may occasionally be waived or replaced by a trustworthy in silico forecast accompanied by one, or better yet, agreement from multiple supplementary software programs. The reasoning behind this solution may be sound, and it may be less expensive than testing. With partially completed data, like the in vitro mutagenicity test, in silico predictions are also a useful tactic. In order to better comprehend a substance's potential to cause DNA damage, a QSAR prediction can be a useful guide prior to conducting the in vitro micronucleus experiment. However, this test is insufficient to assess genotoxicity. According to the ICH M7 guideline, such methods are generally acceptable for the regulatory evaluation of pharmaceutical impurities [10].
3. Genotoxicity Assessment Of Cosmetic Products
In order to identify direct DNA reactive substances that change DNA and, consequently, the genetic code, numerous research teams developed various tests in the second half of the 20th century based on various mechanisms demonstrating direct DNA damages (DNA adduct, unscheduled DNA synthesis, DNA repair chromosomal aberrations). Bruce Ames created the most well-known bacterial reverse mutation test, known as the "Ames test," in the 1970s. Regulatory agencies swiftly considered the most pertinent mutagen tests to detect genotoxic compounds in cosmetics [11], and cosmetics manufacturers used them to optimize their processes and improved ingredients. Regulatory bodies have released test battery strategies for genotoxicity evaluation, and the OECD has produced guidelines.
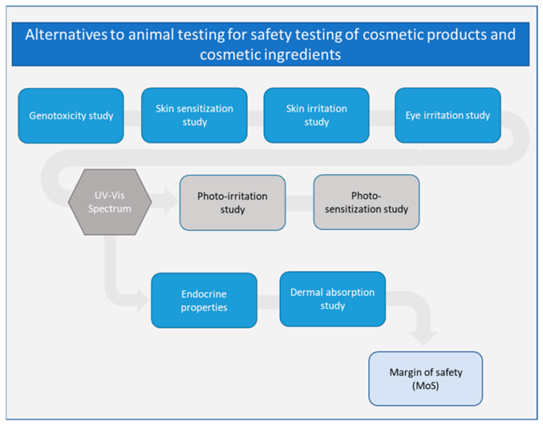
Fig 3.Overview of different alternatives to animal testing for safety assessment of cosmetic products and cosmetic ingredients. Assays in grey are not discussed in this review.
Since the outcome could result in the project's termination, the mutagenicity: bacterial reverse mutation test ought to be carried out initially. The type of test item affects the appropriate approach and, in turn, the anticipated outcome. When using the Ames test on pure chemicals, the test item's structure should be taken into account. Therefore, the metabolic activation system should be modified based on the type of test article (SCCS/1532/14). The Ames test for nanoparticles should be replaced with either the mouse lymphoma assay (OECD 490) or a gene mutation test in mammalian cells (OECD 476). When amino acids are present in complex combinations, like biological molecules or plant extracts, a feeding effect may be seen . The "treat and wash" approach .[12] an in-silico assessment (Quantitative Structure-Activity Relationship QSAR, DEREK, Multicase, or Compound Toxicity Profile) is helpful prior to starting the second genetic toxicology test. The OECD 487 guideline should be followed when performing the micronucleus test in the event of an alarm or when the prediction is out of domain. e. This technique has been improved recently to prevent "false positives." [13]
The compounds known as initiators are identified as a result of this battery of testing. They are carcinogens that react with DNA, as are their metabolites. A second class of compounds, known as promotors in the theory of carcinogenesis, are non-genotoxic carcinogens. To identify genotoxic and non-genotoxic carcinogens, the SCCS/1602/18 (2018) suggests employing the cell transformation assay (CTA) [14] as a novel test substitute for in vivo carcinogenesis investigations.
4. Assessment of Skin Sensitization for Cosmetic Products
Skin sensitizers are substances that possess the inherent ability to cause a hypersensitive reaction in people, potentially leading to allergic contact dermatitis (ACD) after repeated skin exposure. Sensitization activates an adaptive immune response and establishes immunological memory; once this sensitivity develops, it frequently becomes a long-term issue, and the onset of symptoms can only be prevented by avoiding contact with the triggering chemical (see for example [15] for an outstanding review).
Before a new cosmetic ingredient can enter the European market, its safety profile, which includes an evaluation of skin sensitization risks and potency, must be assessed. Following the updates to Annex VII of the REACH regulation and the transition of the cosmetics directive into regulation (EC1223/2009) [1], traditional animal testing methods, like the Guinea Pig tests (GPMT or Buehler test) [16] or the murine Local Lymph Node Assay (LLNA) , are no longer permissible for substances that are solely intended for cosmetic use. In response, numerous New Approach Methods (NAMs) employing in chemico and in vitro strategies have been validated and included in the OECD's official testing guidelines as effective alternatives to animal testing. These methods are focused on specific Key Events (KE) within the Adverse Outcome Pathway (AOP) for skin sensitization [17].
Notably, the empirical data from this publication show that the accuracy of the suggested DAs, ranging from 75.6% to 85.0%, outperforms that of the LLNA (74.2%) in predicting the skin sensitization hazard for humans. Besides the presently recognized OECD assays, numerous innovative and alternative testing methods are undergoing validation and adaptation for recognition as official TGs .[18]
Despite the advancements made to substitute animal testing, further efforts are required to tackle specific challenges associated with existing NAM-based methodologies. For instance, it has been acknowledged that certain chemicals relevant to the cosmetic industry might pose difficulties when assessed using the standard OECD validated tests[19]. The limitations, as they have been identified, are detailed in individual test guidelines (TGs) and may encompass challenges with testing hydrophobic substances, pre-pro haptens, and intricate mixtures like natural extracts, where the ingredient of concern is often present in very low amounts within a complex formulation. Innovative, cutting-edge scientific techniques currently in the OECD Test Guideline Program (TGP) and undergoing review for formal TG adaptation—such as the Genomic Allergen Rapid Detection (GARD) assay [20]
Such discoveries could be beneficial for cosmetic-related testing items, including UVCBs or natural extracts that have low solubility in standard assay solvents like DMSO or water. Moreover, numerous 3D models based on reconstructed human epidermis (RHE) have been developed to help address certain solubility challenges. The majority of these tests have well-defined readouts based on established biomarkers (e.g., IL-18), while some others are less clear.
A recent study assessing the effectiveness of various RHE-based models indicated that most of them displayed comparable, or slightly enhanced, performance (depending on the specific RHE assay) to the top-performing OECD validated test, the h-CLAT assay, when examining a limited range of "challenging-to-test" substances in comparison to human reference data, indicating that such assays could be a valuable source of information within a weight-of-evidence approach for assessments in this chemical area. In addition to the restricted applicability domains, the most apparent drawback of the current OECD validated assays is that they have been validated solely for the identification of skin sensitization hazards, rather than for evaluating the potency of sensitization, which is a vital factor for risk assessment of cosmetic ingredients in consumer products.
Ultimately, as new NAM-based methodologies emerge to replace conventional animal models for the evaluation of cosmetic ingredients, the true measure of these tests' effectiveness in safeguarding human health must be determined by how well they correlate with reliable data regarding the skin sensitizing effects of chemicals in humans. The effectiveness of these methods should not be evaluated solely on how accurately they reflect the shortcomings of traditional "gold" standard animal testing, despite their historical recognition as validated and appropriate OECD approaches. For chemicals that are not yet known to cause sensitization, employing the NAM strategies outlined earlier for the preclinical assessment of cosmetic ingredients is a crucial first step in ensuring the safety profile of cosmetics. Furthermore, as highlighted in [21].
5. Evaluation of the Endocrine Effects of Cosmetics
On December 13, 2017, the European Parliament established scientific criteria to identify endocrine disruptors, which took effect for plant protection products and biocides in 2018 [22]. This represented a significant advancement toward the potential adoption of similar criteria for the regulation of cosmetics within Europe. Although there are differences arising from the unique context of cosmetics, certain lessons about strategies for assessing endocrine properties have been gained from past experiences.
The criteria adopted for identifying endocrine disruptors are closely aligned with the definition provided by the WHO in 2012.[23]
Since 2002, professionals from OECD member countries have released guidelines for testing chemicals focused on endocrine assessment. These globally recognized methods are cataloged, and their appropriate application is outlined in the OECD Guidance Document 150 [24].
Determining an endocrine disruptor ultimately requires clarifying an adverse outcome pathway and necessitates a complete endocrine system for accurate modeling. As noted in the SCCS guidance documents, due to the preservation of endocrine mechanisms across vertebrate species, data from certain ecotoxicological tests may offer valuable insights into the endocrine activity of a compound in humans. This information significantly enhances the weight of evidence available for the endocrine evaluation of cosmetic ingredients. Embryonic forms of aquatic vertebrates serve as ethical and informative models for assessing the endocrine activity of cosmetic ingredients or products within an intact endocrine system. In 2019, the OECD released its first eleuthero-embryo-based test aimed at evaluating thyroid activity, known as Test Guideline 248 (XETA)[25].
While these in vitro aquatic models may not accurately predict effects in humans, they offer a method to identify endocrine activity and serve as a tool for predictive screening. The European Union has adopted hazard-based criteria for evaluating endocrine disruptors. These criteria were applied to regulations for plant protection products and biocides in 2018. Models that illustrate modes of action and associated negative outcomes have supplanted risk assessment in the classification of endocrine disruptors. On the other hand, applying these hazard-based criteria to assess cosmetic ingredients without relying on laboratory animals remains a significant challenge. Nevertheless, some strategies exist to create more realistic exposure scenarios while avoiding regulated life stages of laboratory animals. An approach for screening cosmetics could involve linking the selection of test concentrations for hazard assessments to a variety of daily doses of a compound or product.
6. Evaluation of Skin Penetration of Cosmetic Items
Evaluating dermal absorption is a vital component of ensuring the safety of cosmetic products and their ingredients, unlike drugs that typically enter the body through various routes. In vitro studies on dermal absorption are recognized as the preferred approach for assessing skin pharmacokinetics and are effective in predicting the dermal absorption outcomes in humans. The aim of dermal absorption testing, also referred to as dermal penetration or percutaneous penetration, is to quantify the extent to which a substance passes through the skin barrier and enters the skin itself. Comprehensive guidelines regarding the execution of in vitro skin absorption studies have been provided (OECD 2004, 2011, 2019) [26,27]. Moreover, the Scientific Committee on Cosmetics and Non-Food Products (SCCNFP) established an initial set of "Basic Criteria" for the in vitro evaluation of dermal absorption of cosmetic ingredients in 1999, which was revisited in 2003 (SCCNFP/0750/03) [28]. This Opinion was further updated by the SCCS in 2010 (SCCS/1358/10). Merging the OECD 428 guideline with the SCCS "Basic Criteria" (SCCS/1358/10) is deemed critical for conducting suitable in vitro dermal absorption studies for cosmetic ingredients. Dermal absorption studies are performed to ascertain the extent of a chemical's penetration through the skin and, consequently, its potential for entering systemic circulation. Therefore, understanding dermal absorption mechanisms is crucial for:
Various formulation types can be evaluated through in vitro dermal absorption studies: creams, gels, ointments, suspensions, foams, patches, aqueous solutions, solvents, hair dyes, shampoos, foundations, moisturizers, cleansers, soaps, and sunscreens, among others. Various analytical techniques can be employed to measure the concentration of the test substance in different skin layers, taking into account the physicochemical characteristics of the substance such as lipophilicity, molecular weight, charge, and concentration: these techniques include liquid chromatography–tandem mass spectrometry (LC-MS/MS), inductively coupled plasma–tandem mass spectrometry (ICP-MS/MS), liquid chromatography with UV detection (LC-UV), liquid chromatography with fluorescence detection (LC-Fluo), liquid scintillation counting (LSC) for radiolabelled compounds, and imaging methods like epifluorescence or confocal microscopy for fluorescent molecules or matrix-assisted laser desorption–mass spectrometry imaging (MALDI-MSI) [30].
In vitro dermal absorption testing is highly reliant on the operator, and precautions must be taken particularly when handling skin samples and when removing any excess formulation. The effectiveness of the assay largely depends on the creation and validation of sensitive analytical techniques to measure the concentration of the test substance in the samples. A significant obstacle is how to evaluate dermal absorption in the skin of babies and infants, which is essential for safety assessments of cosmetic ingredients. It is acknowledged that babies, infants, and children constitute a unique subgroup for risk and safety evaluations, and
Researchers routinely take into account the larger skin-surface area relative to body mass in children when conducting safety evaluations of cosmetic ingredients [31].
It is generally believed that systemic exposure in infants and babies is higher than that of older children and adults. On one hand, percutaneous absorption might be increased due to the skin's immature barrier function (a higher skin pH leads to reduced barrier effectiveness and a greater likelihood of irritation), especially in the diaper area.[32]
Conversely, the larger body surface area to mass ratio in babies and infants compared to older children and adults mathematically results in higher dosages in mg/kg bw/w for the same amount of product used[33].
7. Evaluation of Skin and Eye Irritation from Cosmetic Products
Evaluating the potential for skin and eye irritation caused by an ingredient or formulation is a crucial aspect of ensuring the safety of cosmetic ingredients.
Dermal irritation refers to the reversible damage to the skin that occurs after applying a test substance for a maximum of 4 hours (OECD 404) [70].Eye irritation is characterized by observable changes in the eye following the application of a test substance to its anterior surface, with full reversibility within 21 days of application (OECD 405) [34].
RHE is a skin model that consists of living human keratinocytes cultured to create a multi-layered, highly distinct epidermis. The model features well-organized basal cells and includes a functional skin barrier with a lipid profile similar to that found in vivo. RhCE is a corneal model made up of living human cells which are cultured to form a multi-layered, differentiated corneal epithelium. This model also has well-organized basal cells that flatten progressively as the tissue approaches the apical surface, which mirrors the structure of normal human corneal epithelium in vivo. In both models, the cells remain metabolically and mitotically active and release various pro-inflammatory agents (cytokines) that play a significant role in irritation and inflammation. Reconstructed human tissues are cultivated on specialized platforms at the air-liquid interface. The test substance is applied directly to the surface of the tissue, closely simulating “real-life” exposure.
As of now, no in vitro assay or a combination of tests has been validated to serve as a complete substitute for in vivo testing. New testing systems utilizing stem cells are in progress, which may offer novel alternatives for in vitro ocular toxicity evaluation [35].
8. Global Regulatory Responses
Regulatory toxicology is a subset of toxicology focused on safeguarding humans and the environment from the harmful effects of substances through regulations and standardization. Toxicology, often referred to as ‘the science of poisons,’ is a multidisciplinary area of research that examines how chemical, physical, or biological agents can lead to negative outcomes for living organisms and the surroundings. The formal recognition of consumer protection as a governmental responsibility started with the passage of the US Federal Food, Drug, and Cosmetic Act of 1938 [36].
CONCLUSION
The overall number of animal experiments slightly declined in Europe from 2015 to 2017, dropping from 9.59 million to 9.39 million, following a peak of 11.5 million in 2011. The primary use of animals was for research purposes (69%), followed by regulatory needs (23%). In 2017, 61% of animal experiments were conducted for medical products intended for humans, 15% for veterinary products, and 11% for industrial chemicals. Additionally, the European Commission report highlights concerns regarding the use of animals for endpoints where alternative methods are already available, such as irritation and skin sensitization.
Despite the prohibition on the testing of cosmetic ingredients and products on animals, the topic remains contentious. From a regulatory standpoint, the European Agency's position is unequivocal and has been made clearer (“Clarity on interface between REACH and the Cosmetics Regulation”). At present, no cosmetic product undergoes animal testing in Europe. Cosmetic ingredients may rely on previous toxicological test results obtained from animal studies. These results can be derived after the ban on animal testing, provided they are mandated by another regulation (such as food, pharmaceuticals, or even REACH, which addresses worker safety obligations). If a substance's sole application is in cosmetics, then all in silico and in vitro tests will be promoted to establish safety. Nevertheless, for toxicologists, ensuring the absence of risk with the current methods available remains a significant challenge. All so-called New Approach Methodologies, which incorporate AOPs, IATAs, or Defined Approaches, will form the basis for safety assessments of future new ingredients [37].
REFERENCES


Purvesh Patil, Roshan Chaudhari, Sunil Pawar, The Role of Animals in the Safety of Cosmetics, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 3063-3075. https://doi.org/10.5281/zenodo.18700368
 10.5281/zenodo.18700368
10.5281/zenodo.18700368
