M. Pharm Pharmaceutical Chemistry Students Of Al Shifa College Of Pharmacy Kizhattur, Poonthavanam post, Perinthelmanna, Malappuram, Kerala-679325
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used for their analgesic, antipyretic, and anti-inflammatory properties. Despite their efficacy, NSAIDs are associated with significant gastrointestinal (GI) side effects, primarily due to the inhibition of cyclooxygenase-1 (COX-1). This has led to the development of COX-2 selective inhibitors, which aim to reduce GI toxicity while maintaining therapeutic benefits. Prodrugs, compounds that undergo metabolic conversion to release an active drug, have been explored as a strategy to mitigate the adverse effects of NSAIDs. The concept of prodrugs, introduced by Albert in 1951, aims to enhance the pharmacokinetic and pharmacodynamic properties of parent drugs. Recent research has focused on synthesizing prodrugs of mefenamic acid, a widely used NSAID, to improve its therapeutic profile and reduce GI toxicity. Various studies have demonstrated that mefenamic acid prodrugs, such as ester and amide derivatives, exhibit improved anti-inflammatory activity, reduced ulcerogenicity, and enhanced pharmacokinetic properties. These findings suggest that prodrug strategies hold promise for developing safer NSAID therapies with minimized adverse effects. This abstract summarizes the recent advances in the design, synthesis, and evaluation of mefenamic acid prodrugs, highlighting their potential in clinical applications.
NSAIDs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a diverse group of medications utilized for alleviating symptoms such as fever, pain, and inflammation. While their primary mechanism involves inhibiting prostaglandin production by blocking the cyclooxygenase (COX) enzyme, both clinical and experimental evidence suggests that there are additional mechanisms at play.[1] Cyclooxygenase (COX) is the essential enzyme responsible for prostaglandin production. It has two forms: COX-1, which is always present and handles normal bodily functions, and COX-2, which is produced during inflammation. The inhibition of COX accounts for both the therapeutic benefits (through COX-2 inhibition) and the side effects (through COX-1 inhibition) of nonsteroidal anti-inflammatory drugs (NSAIDs). An NSAID that specifically targets COX-2 is likely to maintain strong anti-inflammatory effects while minimizing toxicity.[2] COX-1 is constantly active and produces prostaglandins that protect the stomach and kidneys from damage. COX-2, however, is triggered by inflammatory signals such as cytokines and produces prostaglandins that cause pain and swelling during inflammation. Therefore, selective COX-2 inhibitors should provide anti-inflammatory effects without harming the stomach and kidneys.[3] Upper gastrointestinal (GIT) tract injury is the most common adverse reaction to NSAIDs, frequently leading to GIT ulceration and haemorrhage. The GI mucosal injury caused by NSAIDs is generally attributed to two mechanisms: a local effect from the direct contact of the drug with the gastric mucosa due to the free carboxyl acid group, and a systemic effect following absorption, believed to be due to the inhibition of COX-1, a constitutive form of cyclooxygenase. Intolerance to these GI side effects results in approximately 10% of NSAID users discontinuing the medication. Moreover, users of nonselective NSAIDs are four to eight times more likely to develop gastroduodenal ulcers during treatment.[4]
PRODRUG
The idea of a prodrug was first presented in medicinal chemistry by Albert in 1951. He defined a prodrug as a molecule that does not have inherent biological activity but can convert into a biologically active drug during different stages of its metabolism.[5] Prodrugs are substances that do not exhibit biological activity independently; they need to undergo chemical or enzymatic reactions to become active compounds.[6] Designing prodrugs has been successful in enhancing the physicochemical and biological characteristics as well as the target selectivity of numerous pharmacologically active compounds or drugs.[7] Prodrugs are developed to address issues encountered by the parent drug. They are created to modify pharmacokinetic parameters or physicochemical properties. The objectives of prodrug design can differ. An ideal prodrug should be chemically stable in its formulated dosage form, release the drug at the targeted site, and have a non-toxic promoiety.[8]
PRODRUGS OF NSAIDs
In the past decade, medicinal chemists have focused heavily on the design and synthesis of prodrugs for nonsteroidal anti-inflammatory drugs (NSAIDs). As a therapeutic group, NSAIDs are some of the most widely utilized medications, both by prescription and over-the-counter (OTC).[9] However, they are associated with several unwanted side effects, the most notable being ulcerogenicity, mucosal bleeding, and gastritis.[10] The presence of carboxylic functional groups in NSAIDs is a likely factor contributing to mucosal lining damage.[11] Prodrug design is a strategy used alongside others to address this limitation. The basic idea behind prodrugs is to temporarily block the free carboxylic group found in NSAIDs until they are absorbed systemically.[12] Ester or amide prodrugs of NSAIDs are anticipated to show lower toxicity as they lack a free carboxylic acid group and do not interfere with prostaglandin biosynthesis.[13]
MEFENAMIC ACID
Mefenamic acid, known for its anti-inflammatory and analgesic properties, has been extensively utilized for fifty years. However, with the advent of newer anti-inflammatory and analgesic drugs, the usage of mefenamic acid has substantially declined.[14] Mefenamic acid, like other NSAIDs, hinders the synthesis of prostaglandins, a crucial aspect contributing to its pain-relieving, fever-reducing, and anti-inflammatory effects, which are shared among drugs in this category.[15] Despite being available in different forms such as tablets and suspensions, mefenamic acid's oral administration can cause serious gastric side effects, potentially leading to gastrointestinal bleeding.[16] Utilizing the prodrug strategy could reduce gastrointestinal discomfort linked to mefenamic acid, increase its absorption rate, conceal its unpalatable taste, and extend its duration of effectiveness.[17]
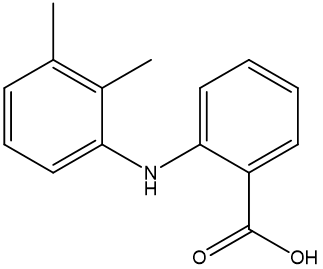
Figure 1 Mefenamic acid
VARIED APPROACHES TO MEFENAMIC ACID PRODRUG DEVELOPMENT
Durga prasad kemisetti et al.,reported Synthesis of Prodrugs of Mefenamic acid and Their in-vivo Evaluation. The prodrug approach is currently a popular method in new drug development. This study focused on developing and investigating prodrugs of mefenamic acid. These prodrugs were successfully synthesized and characterized based on various parameters. Spectral data confirmed that the standard drug had a COOH group as identified by IR and NMR. The structures of PEG 1500/6000-mefenamic acid were verified by IR and NMR, indicating that the COOH group of mefenamic acid had formed an ester, as depicted in Scheme I. Similarly, the structures for PEG 1500/6000-glycine-mefenamic acid were confirmed by IR and NMR, showing that the carboxylic acid group of mefenamic acid had formed an amide linkage with the amino terminal of glycine, as shown in Scheme II. Drug release studies for the synthesized prodrugs, revealed that drug release for PEG 1500-mefenamic acid, PEG 1500-glycine-mefenamic acid, PEG 6000-mefenamic acid, and PEG 6000-glycine-mefenamic acid was higher at pH 7.2 than at pH 1.2. Additionally, the comparison study indicated the significance of the spacer (glycine) in drug release. From the results of the anti-inflammatory activity of the synthesized prodrugs, with measurements taken at 1 hour, 3 hours, and 6 hours for changes in paw volume. Among the four prodrugs, one demonstrated better anti-inflammatory activity than the standard drug. From the ulcer-protecting activity of the synthesized prodrugs. The ulcer index was measured, and out of the four synthesized prodrugs, three offered better protection against ulcers compared to the standard drug. The prodrugs with the best ulcer-protecting activity were PEG 1500-mefenamic acid, PEG 1500-glycine-mefenamic acid, and PEG 6000-glycine-mefenamic acid.[18]
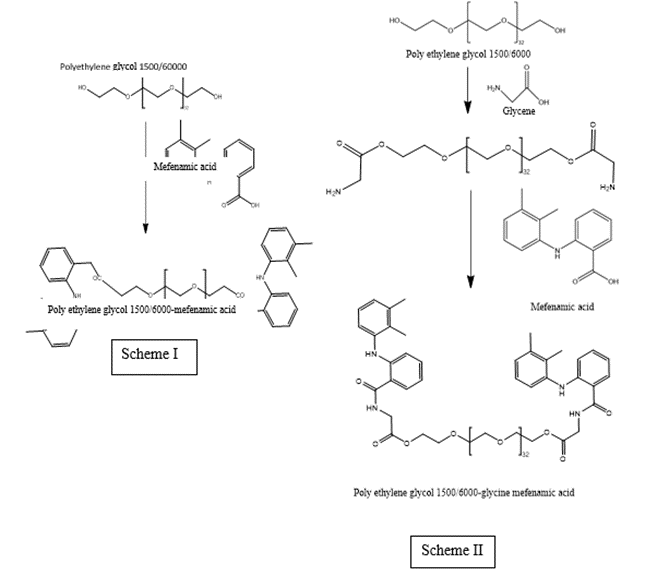
Figure 2 synthesis of prodrugs
Kamal shah et al., reported Synthesis, Kinetics and Pharmacological Evaluation of Mefenamic acid mutual prodrug. A new mutual prodrug (MA-P) combining mefenamic acid (MA) and paracetamol (P) has been created as a gastro-sparing NSAID without ulcerogenic side effects. The structure was confirmed using elemental analysis, infrared spectroscopy, 1H NMR spectroscopy, and mass spectrometry. Ester hydrolysis kinetics were analyzed by HPLC at pH 2, pH 7.4, and in human plasma. The drug's pharmacological activities, including anti-inflammatory, analgesic, and ulcerogenic effects, were assessed. The reduction in ulcerogenicity was evaluated by measuring gastric wall mucosa, hexosamine, and total proteins in the glandular stomachs of rats. The findings indicated that the MA-P ester has a superior ulcer index compared to the parent drug. In this study, the MA-P mutual prodrug was created, synthesized, and assessed as a safer NSAID. The compound proved to be chemically stable and biolabile, maintaining anti-inflammatory effects while significantly reducing ulcerogenicity compared to the physical mixture. This improvement is likely due to better physicochemical properties that enhance bioavailability. These findings suggest that delivering MA and P as a single molecule, the MA-P mutual prodrug, provides clear benefits.[19]

Figure 3 synthesis of mutual prodrug of Mefenamic acid and Paracetamol
Kamal Shah et al., reported Evaluation of mefenamic acid mutual prodrugs. Mefenamic acid-based mutual prodrugs with menthol and thymol have been developed as gastro-sparing NSAIDs, designed to prevent ulcerogenic side effects. The structures of these synthesized esters were confirmed using IR, 1H NMR, and mass spectroscopy. The hydrolysis kinetics of the esters were studied in nonenzymatic buffer solutions at pH 2 and 7.4, and in human plasma using HPLC. Their anti-inflammatory, analgesic, and ulcerogenic properties were evaluated. Additionally, biochemical parameters (GWM and Hexosamine), oxidative parameters (LPO, GSH, CAT, and SOD), and protein levels were analyzed. The findings indicated that the synthesized prodrugs are chemically stable, biolabile, and have optimal lipophilicity. These prodrugs demonstrated a superior ulcer index compared to the parent drug. The prodrugs retained their anti-inflammatory and analgesic properties while significantly reducing ulcerogenicity compared to mefenamic acid. This may be attributed to improved physicochemical properties that enhance bioavailability. Therefore, it can be concluded that administering these prodrugs (mefenamic acid-menthol and mefenamic acid-thymol) offers advantages over administering mefenamic acid alone.[20]

Figure 4 Synthesis of Mefenamic acid – Menthol/Thymol prodrugs
Mina Javanbakht et al., reported Synthesis, characterization and in-vitro evaluation of novel polymeric prodrugs of mefenamic acid. In this study, a series of novel polymeric prodrugs of mefenamic acid were created to reduce its side effects. Initially, glycidyl methacrylate was copolymerized with acrylamide and methyl methacrylate using a free radical solution polymerization method, with ?,?-azobisisobutyronitrile as the initiator at 70±2 °C. Mefenamic acid, a non-steroidal anti-inflammatory drug, was then attached to the copolymers via hydrolysable ester bonds through a transesterification process in the presence of N,N'-dicyclohexylcarbodiimide, resulting in polymeric prodrugs. The structures of these synthesized copolymers and prodrugs were confirmed using FT-IR, 1H NMR, and 13C NMR. These polymeric prodrugs were hydrolyzed in cellophane membrane dialysis bags containing aqueous buffer solutions at pH levels of 1, 7.4, and 8.5 at 37 °C. UV spectrophotometric analysis of the hydrolysis solutions at selected intervals showed that the drug could be released through selective hydrolysis of the ester bond from the drug moiety's side chain. The release profiles indicated that the hydrolytic behavior of the polymeric prodrugs is heavily dependent on the polymer's hydrophilicity, with the release rate of mefenamic acid being higher in alkaline medium than in other conditions. These systems could be useful for developing pH-sensitive polymeric prodrugs for controlled release applications.[21]

Figure 5 Synthesis of Mefenamic acid polymeric prodrugs
J. A. Jilani et al., reported Evaluation of Hydroxyethyl Esters of Mefenamic Acid and Diclofenac as Prodrugs. Hydroxyethyl esters of diclofenac and mefenamic acid were synthesized as potential prodrugs designed to break down easily under enzymatic action. Their stability was assessed in various aqueous buffer solutions with different pH levels (7.4 and 1 N HCl) and in human plasma. While the diclofenac ester showed slow degradation in buffer solutions, with a t1/2 value exceeding 22 hours, it underwent rapid enzymatic breakdown in plasma (t1/2 = 1.12 hours). In contrast, the mefenamic acid ester exhibited greater stability in both buffer solutions (t1/2 exceeding 38 hours at pH 10) and plasma (t1/2 = 7.28 hours) compared to the diclofenac ester. Therefore, the mefenamic acid ester is not considered suitable as a prodrug.[22]

Figure 6 Hydroxy ethyl ester of Mefenamic acid
Bharat V. Dhokchawle et al., reported Synthesis, spectral studies, hydrolysis kinetics and pharmacodynamic profile of mefenamic acid prodrugs. The prodrugs MAEU (mefenamic acid-eugenol ester) and MAVAN (mefenamic acid-vanillin ester) were synthesized using N, N'-dicyclohexyl carbodimide (DCC). The study involved synthesizing mefenamic acid ester prodrugs with eugenol and vanillin using dicyclohexylcarbodimide coupling. Characterization of these prodrugs included Melting Point analysis, Thin Layer Chromatography, Fourier Transform Infrared Spectroscopy (FTIR), Proton Nuclear Magnetic Resonance Spectroscopy (1H NMR), and Mass Spectroscopy to verify their structures. These prodrugs displayed increased solubility in organic solvents, indicating their lipophilic nature, and were determined to be chemically stable and biolabile.The mefenamic acid ester prodrugs demonstrated comparable analgesic and anti-inflammatory effects with reduced ulcerogenicity. This preservation of activity, along with a decline in ulcerogenicity, may be attributed to the analgesic properties of eugenol and its ability to prevent direct contact of the carboxylic group with the gastric mucosa.[23]
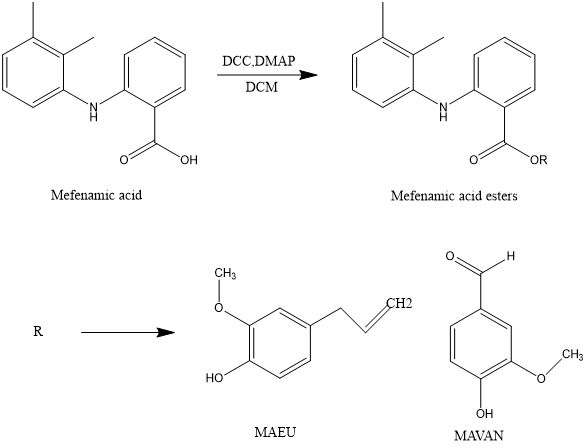
Figure 7 Synthesis of mefenamic acid ester prodrugs with vanillin and eugenol.
Nija B et al., reported Development, Characterization and Pharmacological Investigation of Umbelliferone Conjugates of NSAIDs. The study aimed to develop ester prodrugs of Non-steroidal anti-inflammatory drugs (NSAIDs) such as Mefenamic acid (MA) and Flurbiprofen (FBN) by combining them with the natural antioxidant, 4-methyl umbelliferone. This led to the creation of Mefenamic acid-umbelliferone ester prodrug (MU) and Flurbiprofen-umbelliferone ester prodrug (FU). The main goal was to synthesize prodrugs of NSAIDs with enhanced therapeutic effectiveness and reduced side effects. These prodrugs were synthesized using a coupling method involving N,N’-dicyclohexylcarbodiimide/4 dimethylaminopyrimidine, and were then subjected to comprehensive physical, chemical, and spectral characterization (including IR, 1H NMR, 13C NMR, and Mass spectra), hydrolysis-kinetic study, and pharmacological evaluation for their anti-inflammatory effects, impact on ulcer formation, and influence on degenerative processes in the central nervous system. The study outcomes revealed that upon administration, the umbelliferone-conjugated NSAIDs would release the parent drug via hydrolysis at the intended site, resulting in improved anti-inflammatory activity and decreased gastrointestinal toxicity. Additionally, the synthesized prodrugs exhibited enhanced efficiency in targeting the brain and provided protection against degenerative processes in the central nervous system.[24]
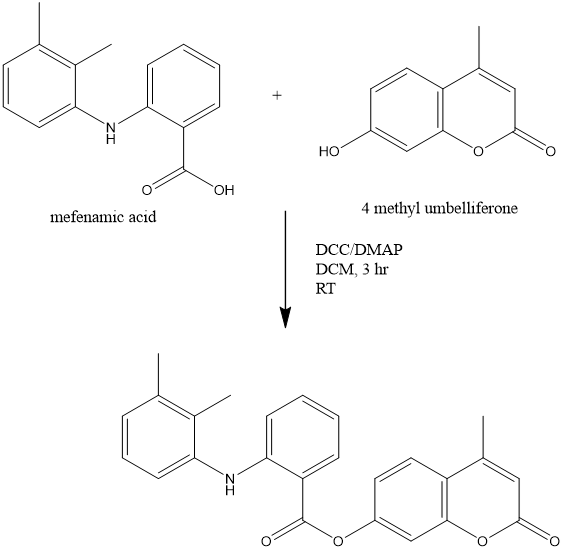
Figure 8 Synthesis of Mefenamic acid - 4 methyl umbelliferone prodrug
Arun Rasheed et al., reported Tyrosine and glycine derivatives as potential prodrugs: design, synthesis, and pharmacological evaluation of amide derivatives of mefenamic acid. This study delves into the synthesis, pharmacological effects, and kinetics of prodrugs derived from mefenamic acid (MA) using tyrosine and glycine. The synthesis process involved a series of protective and deprotective reactions. The hydrolysis of these prodrugs in the intestines was confirmed through kinetic studies in simulated gastric fluid, simulated intestinal fluid, and 80% plasma. The prodrugs were also examined for their analgesic, anti-inflammatory, and ulcerogenic properties. The glycine prodrug displayed the highest analgesic activity at 86%, while both tyrosine and glycine prodrugs exhibited better anti-inflammatory activity at 74% and 81%, respectively, compared to MA's 40%. Additionally, the prodrugs caused fewer gastric ulcers than MA, with tyrosine and glycine prodrugs having average ulcer indices of 9.1 and 4.5, respectively, compared to MA's 24.2. These findings suggest that both prodrugs are more effective than MA and offer the benefit of reducing gastrointestinal side effects.[25]


Figure 9 Tyrosine and Glycine conjugates of Mefenamic acid
CONCLUSION
In conclusion, the prodrug strategy is a versatile and effective approach to overcoming the limitations of traditional NSAIDs like mefenamic acid. By improving pharmacokinetic properties, enhancing bioavailability, and reducing gastrointestinal toxicity, prodrugs hold significant promise for safer and more effective NSAID therapies. Future research should continue to explore novel prodrug designs and their clinical applications to fully realize their potential in improving patient outcomes.
REFERENCE







Neeshma K., Digi Davis C. , Ramsiya K. , Rahila , Razana Binth Yoosuf P. , Rubayyath K. , Shafnaz Abdul Rahman, Revolutionizing NSAID Therapy: The Promise Of Mefenamic Acid Prodrugs, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 7, 306-317. https://doi.org/10.5281/zenodo.12661284
 10.5281/zenodo.12661284
10.5281/zenodo.12661284
