Institute of Pharmacy and Research, Anjangaon Bari Road, Badnera-Amravati, Maharashtra.
Hypertrophic cardiomyopathy (HCM) is a genetic cardiac condition characterized by abnormal thickening of the myocardium, often due to mutations in sarcomere protein genes. This leads to complications like arrhythmias, left ventricular outflow tract obstruction (LVOT), and sudden cardiac death (SCD). Recent advancements have highlighted the importance of early diagnosis using echocardiography, genetic testing, and advanced imaging. Among the therapeutic innovations, Mavacamten, an allosteric cardiac myosin ATPase inhibitor, offers a novel approach by reducing myocardial hypercontractility and improving LVOT gradients. Clinical trials like EXPLORER-HCM have shown significant improvement in cardiac function and patient quality of life with Mavacamten. Furthermore, the role of implantable cardioverter-defibrillators (ICDs) in preventing SCD has been substantiated, especially with the advent of subcutaneous ICDs, which eliminate intravascular lead-related risks. The integration of pharmacological advances like Mavacamten with ICDs provides a synergistic strategy to manage HCM effectively. Despite these advancements, further research is warranted to explore long-term outcomes and optimize therapeutic protocols, particularly in genetically predisposed populations. This review underscores the need for interdisciplinary approaches to enhance HCM treatment and improve patient prognosis.
Hypertrophic cardiomyopathy (HCM) is a heterogeneous myocardial illness that is most usually caused by autosomal dominant sarcomere gene abnormalities. It is the most prevalent monogenic cardiomyopathy in humans. A gene is considered autosomal if it is found on one of the non-sex chromosomes (X or Y), and dominant if the condition is caused by only one mutant copy of the gene. A 50% possibility of inheriting HCM exists for those who have the disorder. The phenotypic manifestations of HCM include left ventricular (LV) hypertrophy, myocardial Hypercontractility, reduced compliance, myofibrillar disarray, and fibrosis. Exertion dyspnoea, chest discomfort, and reduced relaxation and compliance of the left ventricle are frequently caused by microvascular dysfunction, obstruction of the LV outflow tract (LVOT), mitral regurgitation (MR), and other conditions. About half the cases have a family component, whereas the other half have a sporadic component. There is an autosomal dominant form of inheritance. Since penetrance varies with age, many family members may not exhibit the phenotypic when they are examined, leading them to be mistakenly labelled as “unaffected.” Because some family members may acquire HCM later in life, it is vital to periodically evaluate the “unaffected” individuals. In the general population, the estimated prevalence of HCM is 1 in 500. The estimate, however, is predicated on the finding of a less than or equal to 15 mm left ventricular wall thickness in a rather young cohort. Penetrance could lead to a higher true estimate. Many patients may exhibit modest hypertrophy or later experience hypertrophy. The fact that clinical diagnosis is not reliable enough to differentiate the actual HCM from a number of HCM phenocopy disorders may compensate for the underestimate. The most prevalent genes for HCM are MYH7 and MYBPC3, which respectively encode β-MyHC and myosin binding protein-C (MyBP-C) TNNT2, TNNI3, TPM1, and ACTC1 are other frequently occurring genes that encode cardiac troponin T, cardiac troponin I, α-tropomyosin, and cardiac α-actin, in that order. About 60% of all HCM cases have known causative genes, whereas the remaining 40% are still unknown. Genetic testing has little predictive value when significant phenotypic variability is present in HCM.
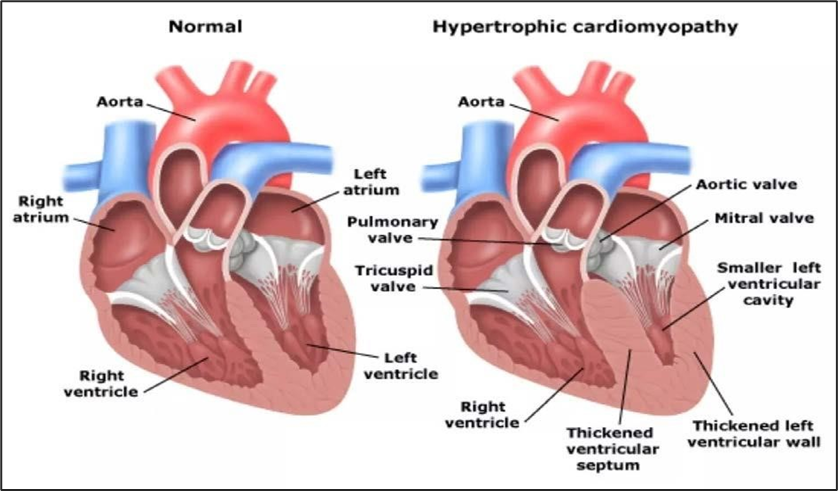
Figure No.1: Normal Heart v/s Heart with Hypertrophic cardiomyopathy
History
Since its modern recognition approximately 50 years ago, hypertrophic cardiomyopathy (HCM) has continued to fascinate the clinicians and the researchers alike. The interest largely has stemmed from the unusual clinical and pathological manifestations of HCM. Our knowledge of the disease has paralleled the development of various medical disciplines and technologies. French pathologist Liouville, who has been credited with the first description of HCM, characterized it as cardiac contraction below the aortic valve in 1869. German pathologist Schmincke described HCM as diffuse muscular ‘hyperplasia’ at the left ventricular outflow tract (LVOT) in 1907. HCM remained a pathological entity until the mid-20th century. The development of advanced diagnostic tools including cardiac catheterization brought in a new phase in our understanding of the disease. Braunwald and colleagues described the dynamic obstruction of the LVOT in a subset of patients with and coined the term idiopathic hypertrophic subaortic stenosis or HSS. Sudden cardiac death (SCD) remains the most dreaded outcome of HCM and often the first presentation of the disease. Clinical studies have shown the effectiveness of automatic internal cardioverter /defibrillators (AICDs) in prevention of SCD, particularly in those at high risk. The challenge is, however, to identify those who are the high risk of SCD. In 1995, Dr. Sigwart introduced percutaneous trans-catheter septal ablation or alcohol septal ablation, which has been shown to be an effective method for reduction of the LVOT obstruction. A recent noteworthy development is the effectiveness of experimental data in prevention and reversal of cardiac hypertrophy in HCM. The findings in animal models of HCM have raised considerable interest in extending the potential beneficial effects of the novel pharmacological agents in treatment and prevention of HCM in human.
Pathogenesis
1.The mechanistic events in HCM might be categorized into 4 sets of interlocking mechanisms.
2.The Primary defect is the mutation. Initial or proximal phenotypes are defined as those resulting from the direct effects of the Mutations on the structure and function of the sarcomere proteins.
3.The intermediary (or secondary) phenotypes include the molecular changes that occur in response to the changes in the sarcomere protein structure and function. Examples of the latter include altered gene expression and activation of the Signalling pathways, such as the MAPK and TGFB1 pathways.
4. The tertiary effects are the ensuing histological and pathological phenotypes, which are the consequence of perturbation of A myriad of secondary molecular events in the myocardium, Such as activation of the hypertrophic signalling pathways. These molecular and histological changes lead to the clinical phenotypes of HCM (quaternary).
5. It is important to note that there is a mechanistic distinction between cases of HCM Caused by sarcomere protein mutation and the phenocopy Conditions because ventricular hypertrophy in the latter may, at least in part, result from storage of material, such as glycogen, and in part because of functional defects in myocytes, Such as impaired contraction.
Symptoms
The majority of people with hypertrophic cardiomyopathy (HCM) have no or few symptoms. In some cases, symptoms are only seen with exercise or exertion. Symptoms may occur during puberty, when hypertrophy develops, but they most commonly start in mid-life. In a smaller percentage of cases, symptoms may not occur until late in life. If symptoms develop, they can vary in severity from one day to another. People with the obstructive form of HCM are much more likely to develop symptoms. The age when symptoms develop and the severity of symptoms vary significantly from person to person. In addition, it is common for symptoms to come and go, with people often reporting "good" (no or little symptoms) days and weeks often separated by "bad"(symptomatic) days and weeks. There is no relationship between a specific gene mutation and the likelihood of developing symptoms or risk for future adverse events.
The most common symptoms include:
? Breathlessness (dyspnoea) on exertion.
? Chest pain may occur at rest or associated with exertion.
? Fainting (syncope) and near-fainting (presyncope).
? Palpitations, awareness of the heart beat or feeling a "forceful" heart beat.
? Light-headedness when sitting or standing up.
? Exertional fatigue or lack of energy (i.e., not having the appropriate exertional stamina for
certain activities).
Diagnosis
1. Echocardiograms – it is often used to diagnose Hypertrophic cardiomyopathy. Sound waves are used to create image of beating heart. This test shows how well the hearts chambers and valve are pumping blood. An Echo also can see if the hearts muscle is thicker than it should be.[4]
2. Electrocardiography (ECG) – This the quick painless test measures the electrical activity of the heart. Sticky patches called electrode are placed on the chest and sometimes the arms and legs. wire connect the electrodes to a computer, which prints or display the test result. An ECG can show irregular heartbeats and signs of heart thickening.
3. Holter Monitor – This small, portable ECG device records the heart activity. it’s worn for a one or two while you do your regular activities.
4. Cardiac CT scan – Rarely, this test is done to diagnose HCM. But it maybe suggested if an MRI can’t be used. a cardiac CT scan uses the X-rays to make pictures of the heart and chest .it can show size of the heart.
5. Genetic testing - Genetic tests are minimally invasive and conducted using a blood or saliva sample. This sample is analysed for 30-50 genes where mutations are linked to HCM. The patient's genes are compared to the genes of normal and HCM-affected patients from a database. Results are usually ready in a few weeks.
Physical Examination
On palpation, the precordial impulse is usually forceful, and Displaced leftward and the peripheral arterial pulses are brisk. In patients with marked ventricular hypertrophy, atrial con-Traction is often especially vigorous, reflected in a prominent Atrial contraction (a) wave in the jugular venous pulse, a pre-Systolic apical lift, and, importantly, a prominent fourth heart Sound (S4). In patients with LVOT obstruction, a harsh, mid systolic, Grade 3 to 4/6 murmur loudest between the apex and the left Sternal border is usually audible. This murmur typically varies with interventions that alter ventricular loading and contractility (see above). Thus, the murmur increases in intensity When left ventricular volume declines during the strain of the Valsalva maneuverer, when assuming the erect position, and during and immediately after exercise. In contrast, it diminishes during squatting, isometric handgrip, and immediately After release of the Valsalva maneuverer. Mitral regurgitation, which is frequently present, is usually accompanied by a Prominent, high-pitched blowing holosystolic murmur loudest at the apex.
Drug profile
Name – Mavacamten
IUPAC Name - (2S)-N-[4-(3,4-dimethoxyphenyl)-1,3-thiazol-2-yl]-2-(4-ethoxyphenyl)-2-(4-methylphenyl)-1,3-benzothiazol-6-amine.
Molecular Formula – C15H19N3O2
Molecular weight – 273.33 g/mol
Structure –
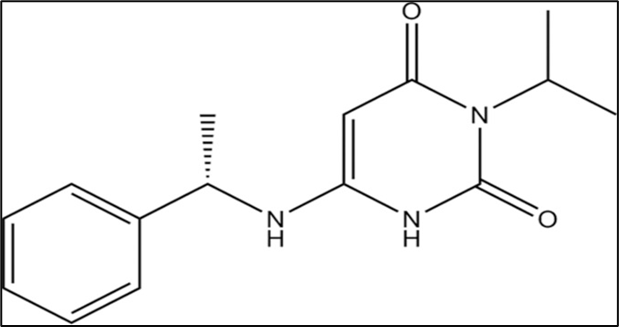
Figure No.2: Chemical structure of Mavacamten.
Category – Cardiac Myosin Inhibitor
Description – White to off- white solid crystal formulation
Solubility – Insoluble in water, but soluble in organic solvents like ethanol, DMSO, and Dimethyl formamide (DMF).
Introduction of Drug
Mavacamten is approved by FDA in April 2022 for adults with symptomatic heart failure (NYHA class II and III) secondary to obstructive HCM. Following the findings of the phase III EXPLORER-HCM trial, which examined the medication’s efficacy in patients with unexplained left ventricular hypertrophy (wall thickness of at least 15 mm) and a peak LVOTgradient of 50 mm Hg or higher at rest, the FDA approved the drug in 2022. Mavacamten was found to improve NYHA cardiac arrest classification, minimize resting and post-exercise LVOT gradient, and enhance exercise performance after 62 weeks. Mavacamten is an entirely novel kind of cardiac myosin ATPase allosteric inhibitor that was created to specifically target the hyper myosin-actin cross-bridge development that is a hallmark of hypertrophic cardiomyopathy. Mavacamten can improve heart filling pressures and lessen blockage of the left ventricular outflow tract (LVOT) by specifically lowering these cross-bridges. This study examines the mechanism of action, pharmacokinetics, dosage, side effects, and contraindications of Mavacamten as well as the value of an interdisciplinary approach to the treatment of patients with obstructive HCM. The US Food and Drug Administration (FDA) has approved Mavacamten, a new oral medicine that is the first of its kind, as an allosteric modulator of cardiac myosin ATPase for the treatment of adult obstructive hypertrophic cardiomyopathy (HCM).
In order to improve functional capacity and lessen symptoms, it is recommended for patients with symptomatic heart failure who are categorized as New York Heart Association (NYHA) class II or III secondary to obstructive HCM.
Clinical Trial Evidence
The EXPLORER-HCM the study showed that Mavacamten-treated patients recovered significantly. Conversely to the much smaller reduction in the placebo group, 57% of patients treated with Mavacamten attained a post-exercise LVOT gradient of less than 30 mm Hg. Assessments of quality of the life, such as the Hypertrophic Cardiomyopathy Symptom Questionnaire Shortness of Breath and the Kansas City Cardiomyopathy Questionnaire Overall Summary Score, showed improvements as well. Following 30 weeks of Mavacamten therapy, decreases in the intracellular myocardial mass index were seen on cardiac magnetic resonance imaging. Using a Markov model, long-term health predictions showed that Mavacamten could offer a gradual improvement in both lifespan and years spent in NYHA functional class I Compared to existing treatments for obstructive HCM.
Mechanism of Action
Mavacamten suppresses cardiac myosin ATPase in a selective, reversible, and allosteric manner. It reduces the prospect of systolic and diastolic cross-bridge formation through lowering the occurrence of actin-myosin cross-bridges. Dysregulation of the relaxed state and excessive myosin-actin cross-bridge production are prevalent in hypertrophic cardiomyopathy (HCM). Mavacamten facilitates the advancement of a super-relaxed, conservation of energy state that reduces obstruction of the left ventricular outflow tract (LVOT) and raises cardiac filling pressures.
Pharmacokinetics
Administration
Strengths and Dosage Forms Available Mavacamten available in dosages of 2.5 mg, 5 mg, 10 mg, and 15 mg as oral capsules. Adult Doses A starting dose of 5 mg once daily is advised. A maximum dose of 15 mg per day can be used for dose titration, which can be done every 4 weeks. The target plasma concentration is from 350 to 700 ng/mL, and it may take several weeks to attain steady-state values. Clinical state, left ventricular ejection fraction (LVEF), and LVOT gradient should be examined before starting treatment and monitored routinely during therapy. Particular Patient Population.
Adverse Effect
Mavacamten might cause a total blockage of ventricular function or aggravate heart failure by reducing systolic contraction. A Reduction in LVEF of up to 10% has been reported.
In the EXPLORER-HCM trial, common adverse effects included dizziness (27%) and syncope (6%).
Other potential adverse reactions are:
Drug -Drug Interaction
Mavacamten exposure is increased when it is used in conjunction with mild CYP2C19 inhibitors or moderate CYP3A4 inhibitors. Starting at 5 mg per day, patients on stable therapy with these inhibitors should begin taking Mavacamten. Starting these inhibitors at a lower dose (e.g., from 15 mg to 10 mg) is recommended for individuals who are currently taking Mavacamten. Because there is no lower dose option for these inhibitors, patients on 2.5 mg Mavacamten should avoid using them. When co-administered with medications that have adverse inotropic effects, including diltiazem, ranolazine, disopyramide, verapamil, or β blockers, should be avoided in order to lower the chance of heart failure and left ventricular systolic dysfunction. When beginning or modifying the dosage of negative inotropic drugs until a steady response is attained, careful monitoring of LVEF is necessary.
Contraindications
Since CYP2C19 and CYP3A4 enzymes metabolize mavacamten, it should not be taken concurrently with strong CYP2C19 or CYP3A4 inducers (like rifampin) or strong CYP2C19 or CYP3A4 inhibitors (like verapamil, ketoconazole). This increases the risk of systolic dysfunction, an aggravation of heart failure, or a loss of medication efficacy. [18]
Toxicity: Overdose Symptoms and Signs
An overdose of mavacamten may result in systolic dysfunction, which can decrease functional ability and cause heart failure. QT prolongation and cardiac osseous metaplasia at high doses (10 mg/kg/day) have been documented in animal investigations.
Discontinuation:
Treatment should be discontinued if LVEF (left ventricular Ejection fraction) falls below 50%. Patients with severe infections Requiring hospitalization or uncontrolled tachyarrhythmia Should be evaluated to determine whether to continue or Interrupt treatment. After discontinuation, an Echocardiographic evaluation should be performed 4 Weeks later. If reinitiating treatment, the dose should be Reduced to the next lower dose (e.g., from 10 to 5 mg Daily), and evaluations at weeks 4 and 8 are Recommended. If LVEF drops below 50% again, Permanent discontinuation is advised.
Implantable Cardioverter Defibrillator (ICD)
For certain patients with hypertrophic cardiomyopathy (HCM), implanted cardioverter defibrillators (ICDs) are recommended for either primary or secondary prevention of sudden cardiac death. Patients with HCM are thought to be more likely to have higher defibrillation thresholds at the time of ICD placement, requiring further steps including lead repositioning, reprogramming, or the insertion of extra ICD leads. Additionally, prior research shows that HCM patients are more likely to experience peri-procedural problems and that a higher frequency of incorrect shocks or failed adequate ICD therapy may exacerbate their clinical course. The largest randomized experiment showing that regular defibrillation is effective was SIMPLE. Defibrillator Testing (DT) during ICD implantation does not decrease mortality or increase shock efficacy. Additionally, individuals receiving DT had higher postoperative troponin levels, which were Linked to poorer long-term results. Considering the outcomes of the SIMPLE ICDs are placed without DT in most cases of the (Shockless Implants Evaluation) experiment. Patients; nonetheless, there is ongoing discussion over the specific need for DT for HCM patients. The introduction of the S-ICD, or totally subcutaneous ICD. Patients who don’t require pacing can benefit from arrhythmia protection with the S-ICD without having to worry about intravascular lead infection or failure. This is especially helpful for young patients with HCM who could avoid the risks of the procedure involved in removing existing transvenous leads as well as the complications that come with them. Patients go through a screening process to make sure their QRS, T-wave morphology, and amplitude are examined in a variety of positions before they are eligible for S-ICD placement. In relation to the QRS complex, if the T-wave is excessively large or delayed.
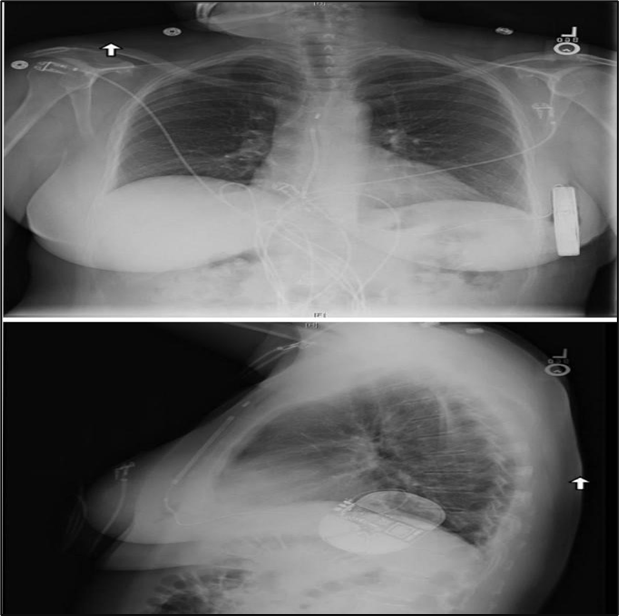
Figure No.3: the subcutaneous implantable cardioverter-defibrillator is implanted in the left lateral thoracic around the fifth and sixth intercostal space and near the mid-axillary line. the defibrillation coil is positioned parallel to the sternum and should ideally be in contact with the deep fascia.
Issues Related to Implantable Cardioverter
The Implantation of Defibrillators before the implantation, a number of concerns must be addressed once a patient is thought to have the potential to benefit from ICD therapy. These include choosing the kind of defibrillator system to use and talking about the dangers of complications from an ICD. Complications associated with ICD are a crucial consideration for patients with HCM, since many are young and will spend a large portion of their adult lives using a device. The patient should be thoroughly informed about two specific risks: the risk of inappropriate ICD therapy and the risk of device-related complications or malfunction. The risk of ICD complications varies from patient to patient due to age and other comorbid conditions. For instance, because of the strain that their growth and development place on leads, children and teenagers seem to have a higher incidence of lead fractures.[34] Additionally, younger patients will need several ICD generator replacements over the course of their lives, raising the possibility of device-related issues. In the HCM population, the reported annual rate of mechanical complications from ICD implantation ranges from 4% to 6%. Pneumothorax, pericardial effusion, haemorrhage, and pocket infection are examples of immediate device complications. Long-term effects like endocarditis and upper extremity venous thrombosis are also included. ICD led performance is becoming more widely known. Research indicates that between 60 and 72 percent of ICD leads are still functional after eight years, and patients who undergo revision are much more likely to experience repeat lead failure or revision. Given that many HCM patients will need reliable, functional transvenous leads for decades, this is especially crucial. Another issue associated with ICD implantation is inappropriate ICD therapy, which is defined as any defibrillation or anti-tachycardia pacing (ATP) administered by the device for circumstances other than persistent VT or VF [34,37]. A recent analysis of a cohort of adult patients with HCM who had ICD implantation detailed the prevalence of inappropriate device therapy [35]. The authors postulated that fewer inappropriate ICD treatments might result from modern ICD programming with longer detection zones and improved supraventricular tachycardia (SVT) discrimination. Despite contemporary ICD programming, the authors discovered that 20% of the cohort’s patients received inappropriate treatment.
CONCLUSION
Hypertrophic cardiomyopathy (HCM) is a genetic heart condition characterized by abnormal thickening of the heart muscle, which can lead to serious complications such as arrhythmias and sudden cardiac death. Early diagnosis through imaging and genetic testing is crucial for effective management, which may include lifestyle modifications, medications, or surgical interventions. Mavacamten has shown promise in treating hypertrophic cardiomyopathy (HCM) by improving cardiac function and reducing symptoms. Its integration with implantable cardioverter-defibrillator (ICD) therapy may enhance patient outcomes by addressing arrhythmia risks associated with HCM. Clinical studies indicate that Mavacamten can potentially reduce the need for ICD interventions, improving quality of life. Research for treatment is essential to fully understand its long-term benefits and safety in conjunction with ICD therapy, making the way for optimized management strategies in HCM patients.
REFRENCES



Yutisha Deshmukh*, Tejashri Kadu, Sachin Dighade, Recent Therapy and Clinical Approaches for Hypertrophic Cardiomyopathy, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 3, 728-738. https://doi.org/10.5281/zenodo.14998473
 10.5281/zenodo.14998473
10.5281/zenodo.14998473
