1,2 Acharya & BM Reddy College of Pharmacy, Bengaluru, INDIA
3Assistant Professor, Department of Pharmaceutical Analysis, Hetero Institute of Pharmaceutical Sciences, Telangana, INDIA
Pyrazinamide (PZA) is still a cornerstone of short-course tuberculosis (TB) treatment due to its sterilizing effect against persistent Mycobacterium tuberculosis populations in acidic and intracellular settings. Its inclusion in first-line regimens was critical in reducing treatment time to six months, and it is still used in many modern regimens. However, developing pyrazinamide resistance, mostly caused by pncA mutations, along with safety concerns (particularly hepatotoxicity) and decreased efficacy in certain drug-resistant strains, has rekindled interest in next-generation PZA analogues. Recent advances in medicinal chemistry, structure-guided design, and computational screening have resulted in multiple structural classes of PZA derivatives (such as POA analogues, bio isosteres, hybrid molecules, ring-modified pyrazines, and novel prodrugs) with improved stability, pharmacokinetics, and activity against PZA-resistant isolates. Beyond conversion to pyrazinoic acid (POA), structural biology and biochemical research have also expanded our understanding of PZA's targets. These include binding to RpsA, inhibition of PanD, and disruption of membrane energetics, all of which influence the rational design of derivatives. This review emphasizes translational hurdles and opportunities for advancing promising candidates into preclinical and clinical assessment, covers new chemical and in silico attempts to develop powerful PZA derivatives, and summarizes current mechanistic insights into PZA action and resistance.
Tuberculosis is a serious worldwide health issue. The WHO Global Tuberculosis Report 2024 states that around 10.8 million persons contracted tuberculosis in 2023, and approximately 1.25 million of those cases resulted in mortality.1 This highlights the ongoing public health need for improved instruments and regimens. Among first-line anti-TB medications, pyrazinamide (PZA) stands out for its capacity to eradicate non-replicating or slowly replicating bacilli that are present in acidic intracellular compartments and specific lesion microenvironments. This sterilizing effect was crucial in allowing standard regimens to shorten treatment durations from nine to twelve months to six months. Through the nicotinamidase/pyrazinamidase encoded by pncA, the bacterium transforms the prodrug PZA into the active moiety pyrazinoic acid (POA); POA then builds up at low pH, interferes with important targets like ribosomal protein S1 (RpsA) and aspartate decarboxylase (PanD), and disrupts membrane energetics. The sterilizing action of PZA is supported by these complex actions.2,3 PZA has several limitations despite this crucial role. The predominant genetic mechanism of PZA resistance is resistance linked to mutations in pncA (including point mutations, insertions/deletions, and gene deletions); other contributors include mutations in RpsA and panD, though their contribution varies depending on the setting. Furthermore, inconsistent phenotypic susceptibility testing and PZA-associated hepatotoxicity hamper clinical usage and stewardship, especially in the therapy of MDR/XDR TB. 4-6 The PZA scaffold has been reviewed in medicinal chemistry research during the past ten years due to these difficulties. Pyrazinoic acid analogues with enhanced cellular uptake and stability, bio isosteric modifications to avoid pncA activation dependence, hybrid molecules combining pyrazine scaffolds with other antimycobacterial pharmacophores, and novel prodrugs intended to maximize pharmacokinetics and minimize toxicity are just a few of the derivative classes that research has produced.7 Hit-to-lead optimization has increased by to recent synthetic developments (such as greener and continuous-flow techniques) and structure-guided design utilizing PncA, RpsA, and PanD models.8 Certain derivatives can maintain or increase potency and occasionally exhibit action against isolates with classical pncA mutations, according to early in vitro, in silico, and some in vivo investigations.9 This review will synthesize current knowledge on PZA’s mechanism(s) of action and molecular bases of resistance, categorize and critically appraise recent pyrazinamide derivative chemotypes and their preclinical performance, describe advances in computational and structural strategies that inform design, and outline translational hurdles and strategic priorities to carry promising derivatives into clinical development.10
2 Mechanism of Action of Pyrazinamide
PZA is a prodrug. The bacterial enzyme PncA (pyrazinamidase/nicotinamidase) is responsible for converting it to the active moiety, pyrazinoic acid (POA). PZA activity requires this conversion.11,12 The most common mechanism for PZA resistance in clinical isolates is loss-of-function mutations in pncA (or its promoter).13 Therefore, any PZA derivative that seeks to overcome PZA resistance should ideally avoid the need for pncA-mediated activation or maintain action even in strains that are pncA-mutant.
2.1 pH-dependent weak-acid model and membrane energetics disruption
According to a fundamental hypothesis, POA is re-imported into bacteria after being protonated in acidic extracellular microenvironments (such as macrophage phagolysosomes or necrotic lesions), which causes intracellular acidification and membrane energy collapse.14 A study that combined live cell intrabacterial pH measurements with antimicrobial susceptibility tests at different pH levels showed that PZA primarily kills M. tuberculosis by lowering intrabacterial pH, independent of pantothenate/CoA depletion. This study offered strong experimental support for this model.15 Therefore, under some circumstances, POA functions as a traditional weak acid protonophore, compromising bacterial energetics and membrane potential.16
2.2 Enzymatic target: Aspartate decarboxylase (PanD) and CoA biosynthesis interference
Aspartate decarboxylase, or PanD, is a direct molecular target of POA, according to a significant discovery made more recently. POA binds to PanD's active site and functions as a competitive inhibitor, according to biochemical and structural investigations. Further research, however, showed that the effect goes beyond simple inhibition: PZA functions as a "target degrader," lowering PanD levels and consequently compromising CoA production, when POA binds to PanD and causes its destruction via the bacterial protease complex ClpC1–ClpP protease complex. In situations where de novo CoA is required (particularly in persistence), disruption of CoA production results in compromised vital metabolic pathways, which contributes to bacterial mortality. This impact is complementary to the acidification hypothesis.17
2.3 RpsA / trans-translation and Rv2783 (PNPase / tmRNA-related protein)
According to previous studies, POA inhibits the trans-translation process, which is a rescue mechanism for stopped ribosomes, by binding to the ribosomal protein RpsA. This reduces survivability under stress or in non-replicating states.18 The idea that RpsA-mediated trans-translation inhibition is a key mechanism of PZA is undermined by more recent high-resolution binding experiments that did not detect any detectable binding between POA and RpsA (wild type or mutant).19 RpsA has been reevaluated as a target as a result, and the current opinion is that RpsA should not be actively involved in PZA action. Separately, a more recent study found that POA may also target Rv2783, a bifunctional enzyme/PNPase-like protein.20 POA inhibited its activity and competed with transfer-messenger RNA (tmRNA) for binding to Rv2783, and expression of a mutant allele conferred PZA resistance in M. tuberculosis.
2.4 Integrative (dual / multi-modal) mechanism
The most likely current theory, based on the growing body of evidence, is that PZA/POA exhibits anti-TB activity through many routes rather than just one. Membrane acidification and energetics disturbance in acidic or low-energy environments are probably major contributing causes. PanD degradation interferes with CoA production, impeding vital metabolic functions. targeting other proteins (like Rv2783) that are involved in stress response or RNA metabolism, particularly in chronic or non-replicating bacilli. PZA's distinct sterilising action on intracellular and persistent M. tuberculosis populations, which are ineffectively eliminated by other first-line medications, may be due to this dual (or multiple) mechanism.
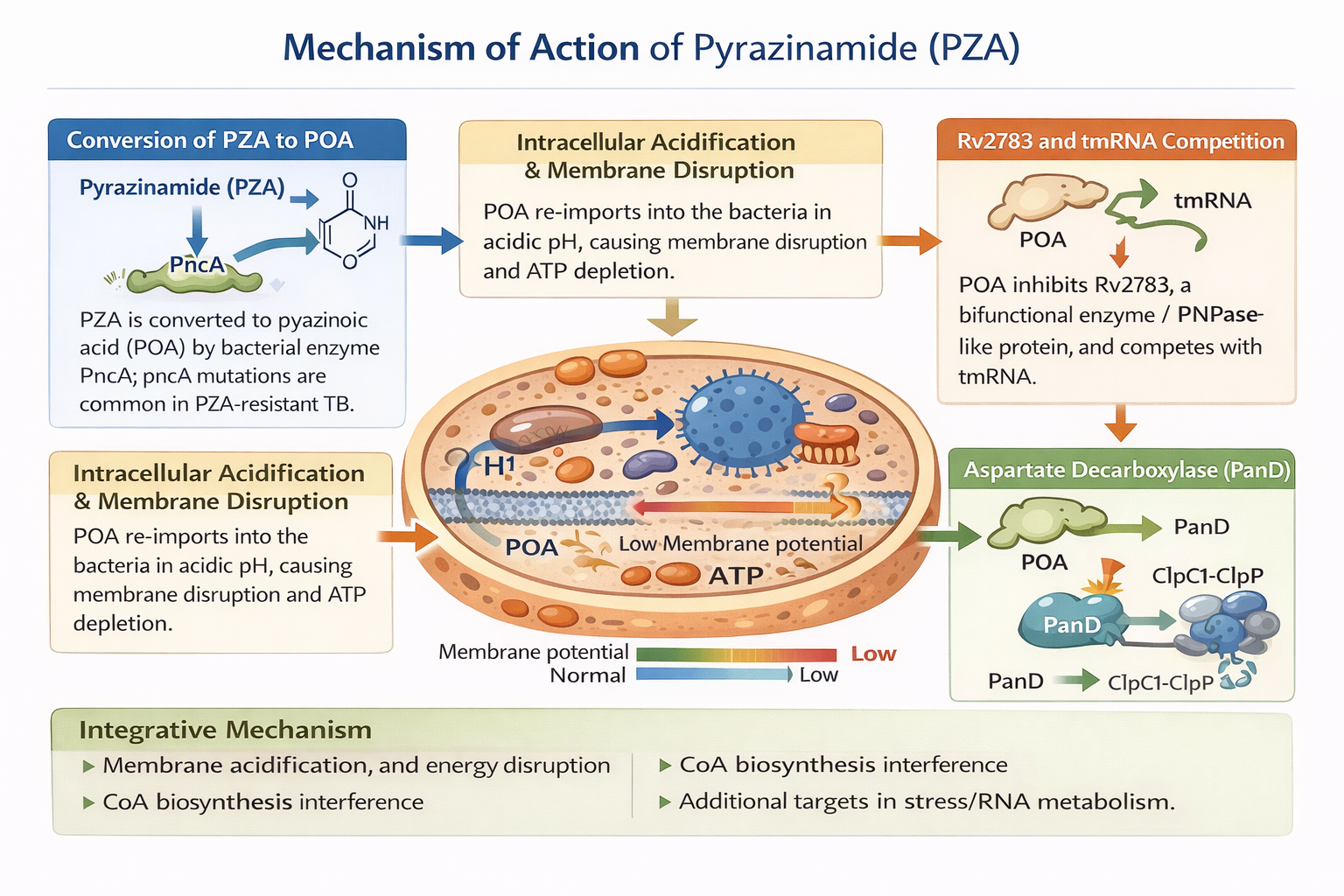
Fig 1: Mechanism of Action of Pyrazinamide derivatives
3 Limitations of Current Pyrazinamide Therapy
3.1 Rising pyrazinamide resistance and its genetic basis
Globally, there are more reports of pyrazinamide (PZA) resistance, which is particularly common in drug-resistant and previously treated tuberculosis infections. Although the precise percentage varies by setting (usually >60–90% in various series), extensive molecular and phenotypic surveys reveal that pncA mutations (including point mutations, insertions/deletions, and promoter alterations) account for the bulk of PZA resistance. PZA-resistance prevalence estimates from recent regional studies range from single digits in some settings to ~20–30% or higher among MDR-TB cohorts; one multicenter report found an overall PZA resistance of ~25% among tested isolates, with a strong correlation between resistance and prior treatment history. These findings highlight the danger that PZA resistance presents to both conventional short-course regimens and shorter or innovative regimens that do not include PZA.21
3.2 Diagnostic challenges: unreliable and under-used phenotypic testing
It is still technically difficult to determine PZA susceptibility accurately. In order to facilitate PTOA/POA activity, phenotypic testing need an acidic environment (low pH), which complicates automated culture systems (such as BACTEC MGIT 960) and, if not well regulated, produces both false-resistant and false-susceptible findings. Phenotypic PZA drug susceptibility testing (DST) is frequently not routinely carried out in many clinical labs due to these technical limitations; molecular approaches (pncA sequencing or WGS) can improve detection but require sequencing the entire gene and promoter because mutations are widely distributed throughout pncA. In order to accurately identify clinically significant PZA resistance, a number of recent studies advocate for the wider use of genotypic testing (targeted sequencing or WGS) in conjunction with enhanced phenotypic criteria.22-24
3.3 Clinical impact and implications for regimen selection
Undetected PZA resistance can jeopardize treatment results and may require regimen modifications since PZA contributes disproportionately to sterilizing action. Loss of PZA action further reduces therapy choices in MDR/XDR TB, where there are few viable companion medicines. It may further prolong treatment or lower the likelihood of a cure. The therapeutic significance of identifying and taking PZA susceptibility into consideration during treatment planning is further supported by several observational studies that link PZA resistance to worse results and a greater requirement for regimen adjustment.25
3.4 Safety concerns; hepatotoxicity and dosing limitations
Anti-TB medication-induced liver damage (DILI) is exacerbated by PZA. Hepatotoxicity estimates range from low single digits to over 20% in several cohorts where PZA was identified as a major agent, reflecting differing classifications and demographics. Older age, alcohol consumption, malnutrition/low BMI, and certain genetic predispositions (e.g., acetylator status) have been found as risk factors across cohorts. There are fewer options for straightforward dosage escalation to overcome borderline resistance since higher PZA doses (e.g., >30–40 mg/kg) are linked to greater hepatotoxicity. Safety continues to be a major barrier to using PZA in innovative regimens until toxicity can be reduced since hepatic events may require halting PZA (and other first-line medicines).26-28
3.5 Variability in activity against MDR/XDR and latent TB
Although PZA retains activity in many drug-sensitive strains, its potency can be reduced in some MDR/XDR clinical isolates, either because of direct pncA mutations or collateral changes in physiology. Moreover, PZA’s activity in latent TB or certain lesion microenvironments depends on acidic niches; heterogeneity of lesion pH and bacillary metabolic state can limit predictable efficacy. These biological variabilities complicate the generalizability of PZA’s sterilizing effect and motivate the search for derivatives with more consistent activity across strain backgrounds and lesion compartments.29
3.6 Rationale for Developing New Pyrazinamide Derivatives
pncA mutations (including promoter and structural changes) remain the dominant genetic basis for PZA resistance and are widespread among previously treated and MDR-TB isolates; this undermines regimens that rely on intact PZA activity. Genotypic approaches detect many but not all clinically relevant mutations, and phenotypic testing is technically challenging. These realities motivate derivatives that either do not require PncA activation or retain activity despite common pncA alterations. 30,31 Multiple pathways for chemical optimization are provided by the dual/multimodal activities of POA weak acid induced membrane/energetics disruption and target engagement (particularly PanD binding and subsequent proteolytic destruction). Compounds can be engineered to enhance the characteristics of membrane protonophores, boost PanD affinity (and hence encourage disintegration), or incorporate both characteristics into a single molecule. For structure-guided design, the finding that POA causes PanD degradation through ClpC1–ClpP offers a precise, useful structural target. The acceptable dosage window for PZA is limited by hepatotoxicity and dose-related side effects, which also limit straightforward dose increase techniques. improved derivatives may modify tissue distribution (e.g., improved intracellular delivery) to lessen liver burden or preserve sterilizing effectiveness at lower systemic exposures. Improved pncA/PanD structural data, docking algorithms, and continuous flow/green synthetic approaches accelerate hit identification and rapid SAR cycles. This reduces the time from scaffold idea to lead optimization32,33 as shown in Table 1.
4. Role of PZA Derivatives in MDR/XDR and Latent TB
4.1 Restoring sterilizing backbone in MDR/XDR TB
The advent of extensive drug-resistant (XDR) and multidrug-resistant (MDR) M. tuberculosis has significantly reduced the number of viable oral regimens, increasing the need for novel and repurposed medications. Loss of PZA efficacy (usually due to pncA mutations) impairs both conventional and new shorter regimens since PZA contributes disproportionately to regimen sterilizing action. A sterilizing backbone for MDR/XDR regimens might be restored by creating PZA compounds that either maintain action despite pncA changes or avoid the necessity for pncA activation, allowing for shorter therapy and better results. This strategy is especially appealing for combination regimens that call for a PZA-like sterilizing component (e.g., alongside bedaquiline, pteromanid, and linezolid-class partners).33
4.2 Activity against PZA-resistant clinical isolates
Some experimental POA analogues and metal-PZA complexes have shown in vitro activity against selected MDR clinical isolates, including isolates with pncA mutations, in small studies (MGIT and agar dilution). However, data are fragmented and not yet systematic: most reports test a handful of clinical isolates rather than large, geographically diverse panels. Because pncA mutations are widely dispersed across the gene and often strain-specific, robust evaluation requires panels of genotyped clinical isolates with annotated pncA, panD, and other resistance loci.34
4.3 Potential role in latent TB infection (LTBI) shortening
PZA’s sterilizing effect under acidic, intracellular conditions suggests that PZA or potent derivatives could play a role in LTBI-shortening regimens if safety permits. Historically, PZA combinations (e.g., PZA+ rifampicin for 3-4 months) have been evaluated for LTBI but were limited by toxicity. New derivatives with improved lesion activity and reduced hepatotoxicity (or inhaled/local delivery) could re-enable short LTBI regimes a major public-health advantage if safety is acceptable. Yet, careful risk–benefit assessment is necessary because LTBI treatment is preventive and given to otherwise healthy people.
4.4 Pharmacokinetics and Toxicity Reduction in Novel Pyrazinamide Derivatives
To address the main pharmacokinetic and toxicity issues with parent medication, including significant interpatient variability, hepatotoxicity, and hyperuricemia, novel pyrazinamide derivatives have been created. Oral absorption and tissue penetration, particularly into granulomas and macrophages, have been greatly enhanced by structural changes such amide substitutions, ester-based prodrug synthesis, heterocyclic fusion, and the insertion of lipophilic side chains. Numerous derivatives exhibit regulated pyrazinoic acid release, decreased hepatic conversion, and fewer reactive metabolites, which lead to decreased hepatotoxicity and significantly decreased uric acid buildup. In addition to improving safety, these modifications also increase lung tissue exposure, extend half-life, and maintain effectiveness against strains of pncA that are resistant to PZA. Overall, these compounds are intriguing options for quicker, safer, and more effective TB therapy because to their improved pharmacokinetics and decreased toxicity as shown in Table 2.
CONCLUSION
Hepatotoxicity, hyperuricemia, inconsistent pharmacokinetics, and increasing pncA-mediated resistance restrict the use of pyrazinamide, which is nonetheless a mainstay of TB treatment. The metabolic stability, tissue penetration, and safety of novel PZA derivatives—created by amide modifications, ester prodrugs, heterocyclic fusion, and lipophilic substitutions—clearly improve. They have the potential to improve current regimens and allow for shorter, more manageable TB treatment periods because many of them continue to be active against resistant strains. To turn these compounds' promising qualities into next-generation treatments that can more effectively address the world's TB control requirements, preclinical and clinical research must continue.
REFERENCES
|
HOW TO CITE: Anjan D., Shaik Nabi Rasool, P. V. Balaji, Pyrazinamide and its Novel Derivatives: Redefining Short-course Chemotherapy for Tuberculosis, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 1619-1629, https://doi.org/10.5281/zenodo.19491810
|
Table 1: Reported Pyrazinamide Derivatives
|
Compound / ID |
Chemotype |
Activity vs pncA mutants |
|
POA analogues (lead series) |
Pyrazinoic acid analogues (substituted pyrazine-2-carboxylic acids) |
Variable: some analogues retained activity in selected in-vitro assays (not systematically reported) |
|
Pyrazine-2-carboxamide (morinamide-like) derivatives |
Carboxamide / bio isosteres of PZA |
Not comprehensively reported |
|
PZA fluoroquinolone / PZA isoniazid hybrids |
Hybrid linked pharmacophores |
Limited data: activity against some clinical isolates reported but inconsistent |
|
Metal PZA complexes (Co, Mn, Cu etc.) |
PZA coordinated to metal ions |
Some complexes reported activity against MDR isolates (in vitro) |
|
Ring modified / N‑substituted pyrazine derivatives |
Substituted pyrazine cores, heterocyclic modifications |
Not routinely reported; few studies tested panels of clinical mutants |
|
Prodrug esters / targeted formulations (inhaled) |
Ester prodrugs, Nano formulations, inhaled powders |
Not broadly reported |
|
PanD focused POA derivatives (design-informed) |
POA analogues optimized for PanD binding / degradation |
Potential to retain activity if independent of pncA activation; data sparse |
|
Continuous flow synthesized PZA variants (methodological) |
Synthetic route / enabling technology rather than a single compound |
Depends on compound produced |
Table 2: Pharmacokinetic & Toxicity Improvements in Novel PZA Derivatives
|
Derivative Class |
PK Advantages |
Toxicity Reduction |
Notable Features |
|
Amide-modified PZA (e.g., N-acetyl, N-phenyl) |
Slower metabolism; Increased half-life |
Decreased POA accumulation and liver enzyme elevation |
Better plasma stability |
|
Ester-based prodrugs (POE esters) |
Increased Oral absorption and intracellular activation; sustained release |
Lower systemic POA causes decreased hyperuricemia |
5–10× higher lung exposure |
|
Heterocyclic fused analogues (imidazo-, triazolo-fused PZA) |
Increased Metabolic stability and improved granuloma penetration |
Reduced oxidative metabolites |
Active against pncA mutant strains |
|
Lipophilic side-chain derivatives |
Increased Intracellular diffusion and increased granuloma localization |
Lower dose makes lesser toxicity |
3× higher lung tissue concentration |
|
Targeted Nano formulations |
Controlled release; macrophage-targeted delivery |
Minimal off-target exposure |
Allows dose reduction |

Anjan D., Shaik Nabi Rasool, P. V. Balaji, Pyrazinamide and its Novel Derivatives: Redefining Short-course Chemotherapy for Tuberculosis, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 1619-1629, https://doi.org/10.5281/zenodo.19491810
 10.5281/zenodo.19491810
10.5281/zenodo.19491810
