P. Rami Reddy Memorial College of Pharmacy, Kadapa, Andhra Pradesh, India.
A post-zygotic mosaic mutation in the AKT1 gene causes the uncommon, progressive, and sporadic overgrowth disorder known as Proteus syndrome. Its defining feature is asymmetric tissue proliferation involving the skin, adipose tissue, connective tissue, and bone. Characteristic clinical manifestations include cerebriform connective tissue nevi, skeletal abnormalities, vascular malformations, and an increased risk of tumor development. Diagnosis is established through clinical criteria, imaging, and genetic confirmation from affected tissue. Management requires a multidisciplinary approach, including surgical correction, supportive care, anticoagulation, and emerging targeted therapies. We report the case of a 42-year-old man who presented to the Department of Dermatology, Venereology, and Leprosy, Government General Hospital, Kadapa, with complaints of progressive swelling of the left hand and difficulty lifting weights for one year. He also noted the development of multiple, verrucous hyperpigmented lesions over both hands and legs for the past month. Clinical evaluation revealed asymmetric overgrowth with verrucous skin lesions. Radiological studies confirmed skeletal hypertrophy, while dermatological findings were consistent with epidermal nevi. Proteus syndrome was diagnosed based on clinical features and supportive investigations, with multidisciplinary management instituted to optimize outcomes. This case underscores the importance of early recognition and highlights the rarity of adult-onset presentations
Proteus syndrome (PS) is an ultra-rare, sporadic overgrowth disorder characterized by progressive, segmental, and asymmetric proliferation of multiple tissues, most commonly bone, skin (notably cerebriform connective-tissue nevi), adipose tissue, and the central nervous system [1,2]. Clinical signs are typically absent at birth and emerge between 6–18 months of age, then accelerate through childhood, causing disproportionate, patchy deformities and functional morbidity [2,3]. The condition is caused by post-zygotic (somatic) mosaic activation of AKT1 classically the p.E17K variant driving PI3K–AKT pathway hyperactivation and explaining the mosaic distribution of lesions [1]. Complications include vascular malformations, thromboembolism (a leading cause of early mortality), orthopaedic deformities, pulmonary involvement (including bullous lung disease), and a predisposition to certain benign and malignant neoplasms; care is multidisciplinary and risk-based [3,5]. PS is exceedingly uncommon (estimated <1 per 1,000,000), with only a few hundred cases reported worldwide, and remains a clinic-molecular diagnosis supported by characteristic phenotype plus detection of AKT1 mosaicism in affected tissue [2,6].
CONCLUSION:
Proteus syndrome is an exceptionally rare, progressive disorder characterized by post-zygotic mosaic mutations in the AKT1 gene, leading to highly variable phenotypic expression and significant diagnostic challenges. Although the condition typically manifests during early childhood, our report underscores that adult-onset presentations, though uncommon, can occur and may result in diagnostic delay. Establishing an accurate diagnosis requires a high index of clinical suspicion, supported by thorough physical examination, advanced imaging modalities, and, where available, confirmatory molecular testing. Given the disorder’s multisystem involvement and the potential for life-threatening complications including thromboembolic events, skeletal deformities, and neoplastic transformation comprehensive, multidisciplinary management is imperative. Early recognition and individualized therapeutic strategies not only mitigate morbidity but also substantially improve functional outcomes and quality of life. Furthermore, continued documentation of rare adult presentations is essential to broaden the current understanding of the natural history of Proteus syndrome, refine diagnostic algorithms, and contribute to the development of targeted molecular therapies.
CASE REPORT:
History
A 42-year-old man presented to the Department of Dermatology, Venereology, and Leprosy, Government General Hospital, Kadapa, with complaints of abnormal swelling of the left hand and difficulty lifting weights for the past one year. He also noticed multiple raised, hyperpigmented verrucous lesions over both legs and hands for one month. The swelling was progressive and painless, associated with functional limitation. There was no history of trauma, drug intake, systemic illness, or relevant family history.
Examination
General examination was normal. Local examination revealed asymmetrical hypertrophy of the left hand with Marked deformity and restricted movement. Multiple verrucous, hyperpigmented, raised cutaneous lesions were present over the upper and lower limbs, consistent with epidermal nevi. The overgrowth was firm on palpation, non-tender, and without local rise of temperature. No neurological deficits or systemic organ involvement were detected on clinical evaluation.
Investigations
Histopathological examination of verrucous lesions from the leg and hand revealed hyperkeratosis, acanthosis, and papillomatosis with elongated rete ridges and focal basal layer hyperpigmentation. The dermis showed irregular connective tissue proliferation, adipose tissue hyperplasia , and ectatic vascular channels. These findings were consistent with Proteus syndrome.
Diagnosis
Based on the characteristic clinical features of asymmetric limb overgrowth, verrucous epidermal nevi, and progressive deformity, supported by histopathological findings of connective tissue proliferation, adipose tissue hyperplasia, and vascular malformations, a diagnosis of Proteus syndrome was established.

Figure:1 Macrodactyly and asymmetric overgrowth of the toes
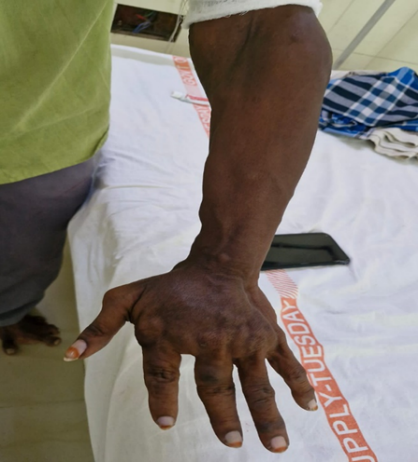
Figure:2 asymmetric hypertrophy and deformity of the left hand
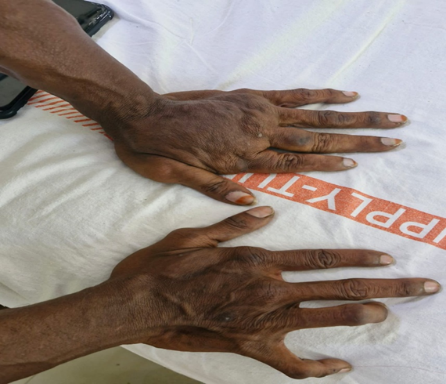
Figure:3 depicting asymmetric hypertrophy and deformity of the hands
Treatment
The patient was initiated on cefixime 200 mg (antibiotic for secondary bacterial infection), pantoprazole 40 mg (proton pump inhibitor for gastric protection), paracetamol 500 mg (analgesic for pain control), and topical fusidic acid (for cutaneous lesions). This regimen was maintained during hospitalization (7 days)
Outcome
The patient was managed with symptomatic and supportive measures under a multidisciplinary team. Swelling and functional limitation of the left hand persisted, but cutaneous lesions were stabilized with dermatological care. The patient remains under regular follow-up for monitoring of progression and systemic complications.
DISCUSSION:
Proteus syndrome is caused by post-zygotic activating mutations in AKT1, leading to mosaic hyperactivation of the PI3K–AKT pathway and asymmetric tissue overgrowth [1,2]. Management is challenging due to variable presentation, risk of thromboembolism, and progressive multisystem involvement, necessitating a multidisciplinary approach [3,4]. Surgical correction, anticoagulation, and supportive care remain the mainstay of treatment, although with frequent recurrence and significant morbidity [3]. Emerging targeted therapies such as the AKT inhibitor miransertib show promise in modifying disease progression and improving outcomes [5].
Etiology and Pathogenesis
Proteus syndrome is caused by a post-zygotic somatic activating mutation in the AKT1 gene, most commonly the p.E17K variant, which results in mosaic distribution of affected tissues [1]. This mutation leads to constitutive activation of the PI3K–AKT signaling pathway, promoting abnormal cellular proliferation, survival, and tissue overgrowth [1,2]. The mosaicism explains the patchy and asymmetric involvement, distinguishing Proteus syndrome from other generalized overgrowth disorders[13]. Phenotypic variability is marked, ranging from mild cutaneous lesions to severe skeletal deformities and visceral involvement [3,9].
Management Challenges and Approaches
Management of Proteus syndrome is complex due to its rarity, phenotypic heterogeneity, and progressive course[12].Challenges include delayed recognition, difficulty distinguishing it from other overgrowth syndromes (such as CLOVES or Bannayan-Riley-Ruvalcaba syndrome), and the high risk of morbidity and mortality. Thromboembolic events represent the leading cause of premature death in these patients [4]. Current treatment is largely supportive and symptomatic, requiring a multidisciplinary approach involving dermatology, orthopedics, vascular surgery, pulmonology, oncology, and rehabilitation medicine[3,7]. Interventions include surgical correction of skeletal deformities, excision of disfiguring or symptomatic lesions, anticoagulation for thrombotic risk, and vigilant surveillance for neoplasia[10,11].
Emerging Therapies: Biologics
Recent advances in molecular genetics have paved the way for targeted therapies. The identification of AKT1 activation has led to the trial of AKT inhibitors, such as miransertib (ARQ 092), an oral allosteric pan-AKT inhibitor, which has shown early promise in reducing overgrowth and improving symptoms in Proteus syndrome and related mosaic overgrowth disorders [7,15]. Other agents targeting the PI3K–AKT–mTOR pathway are under investigation, offering hope for disease-modifying therapy. However, access, long-term efficacy, and safety remain areas of ongoing research[14].
CONCLUSION:
Proteus syndrome is an exceptionally rare, progressive disorder with complex clinical challenges. This case emphasizes the possibility of adult-onset presentation and underscores the importance of early recognition, multidisciplinary care, and vigilant monitoring to reduce morbidity and improve outcomes.
ACKNOWLEDGEMENT:
We sincerely thank the patient and his family for their cooperation and consent in reporting this case. We also acknowledge the contributions of the multidisciplinary team at the Department of Dermatology, Venereology, and Leprosy, Government General Hospital, Kadapa, for their valuable clinical support and management.
CONFLICT OF INTEREST: the authors declare that there is no conflict of interest.
ABBREVIATIONS:
PS-Proteus Syndrome, AKT1-v-AKT Murine Thymoma Viral Oncogene Homolog 1, PI3K Phosphoinositide 3-Kinase, mTOR -Mammalian Target of Rapamycin, PPI - Proton Pump Inhibitor, HPE -Histopathological Examination.
PATIENT CONSENT: The patient mentioned is this case report has given permission for it to be published understanding, that there identify will kept private and accepting nature of the report. The patient is happy with the prescription drug they were given.
SUMMARY:
Proteus syndrome is an extremely rare and sporadic overgrowth disorder resulting from a somatic activating mutation in the AKT1 gene, leading to mosaic dysregulation of the PI3K–AKT pathway. It is characterized by progressive, asymmetric proliferation of multiple tissues, particularly bone, skin, adipose, and connective tissue. Distinctive clinical features include cerebriform connective tissue nevi, skeletal deformities, vascular malformations, and epidermal nevi, with an increased risk of neoplasia. The disorder typically manifests in early childhood, progresses throughout life, and is associated with significant morbidity. Diagnosis is established through clinical criteria, imaging studies, and confirmation of AKT1 mosaicism in affected tissue. Management remains multidisciplinary and supportive, with surgical correction, anticoagulation, and rehabilitative measures forming the mainstay of care. Targeted therapies, such as AKT inhibitors, represent a promising area of research for altering the natural course of the disease.
REFERENCES


C. Lavanya Thejonidhi, Y. Bhargavi, M. Dinesh, Proteus Syndrome Presenting with Multisystem Involvement: An Uncommon Case Report, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 2923-2928. https://doi.org/10.5281/zenodo.16980399
 10.5281/zenodo.16980399
10.5281/zenodo.16980399
