Research Scholar, Siddhi’s Institute of Pharmacy, Nandgoan Murbad Thane 421401.
Pharmacovigilance is defined as the processes of monitoring and evaluating ADRs, and it is an essential component of effective drug control systems, clinical practice, and public health programs. As the number of reported adverse drug reactions (ADRs) has increased, pharmacovigilance requires skills to identify potential risks and its removal. ADRs are a leading source of morbidity and mortality, mostly found through post-marketing surveillance. Improving ADR reporting in India, includes using novel methods like Pharmacovigilance, is critical for enhancing patient safety and public health outcomes. Active pharmacovigilance surveillance involves monitoring drug events, adverse events, and reporting them spontaneously. The monitoring of adverse drug responses, which begins with the pre-marketing of new medications and continues through the post-marketing phase of medications, is significantly influenced by pharmacovigilance. Given India's huge population, diverse illness frequency patterns, and traditional medical practices, it is important that the country implement strong pharmacovigilance programs. All the regulatory authorities, institutions even clinicians and physicians are initiating nation-wide pharmacovigilance programmes to collect, collate, analyse data on adverse drug reactions for protecting the health of the patients. The ultimate objective of this review is to understand role of pharmacovigilance and make sure that the advantages of medication use outweigh risk factors in order to protect health of Indian population.
Pharmacovigilance is defined as the science of detection, assessment, and prevention of adverse drug reactions in humans. Under-reporting of drug reactions is the major problem and has various reasons. The WHO has initiated the program of reporting all adverse drug reactions now coordinated by the Uppsala Monitoring Centre in Uppsala, Sweden, with oversight by an international board. This review presents in brief the relevance, functioning, importance, and the procedure of reporting adverse drug reactions and how pharmacovigilance plays major role in it. Pharmacovigilance has been confined, mainly to detect adverse drug events that were previously either unknown or poorly understood. Pharmacovigilance is an important and integral part of clinical research and these days it is growing in many countries. Today many pharmacovigilance centers are working for drug safety monitoring in this global pitch, however, pharmacovigilance faces major challenges in aspect of better safety and monitoring of drugs.[1]
History of pharmacovigilance:
Earlier, very few novel drugs were discovered in India and hence the need for pharmacovigilance system was less. However, India is now rapidly progressing in the field of drug discovery. There is also rapid increase in the marketing of foreign branded drugs. Such developments have led to the emergence of a strong pharmacovigilance system in our country. Pharmacovigilance system in India is regulated by Schedule Y of the Drugs and Cosmetics Act 1940, and Rules, 1945. This Schedule contains all specifications for conducting clinical trials on animals and humans for the development of a new drug and requirements of clinical trial for the import, manufacture and approval of marketing of a new drug in India. The Schedule contains a section on post-marketing surveillance which outlines the details about the format to be followed for Drafting periodic Safety Update Reports (PSURs), their submission and PSUR cycle. It also contains time frames in which these reports should be submitted to the concerned authorities. Unexpected and serious adverse effects in clinical
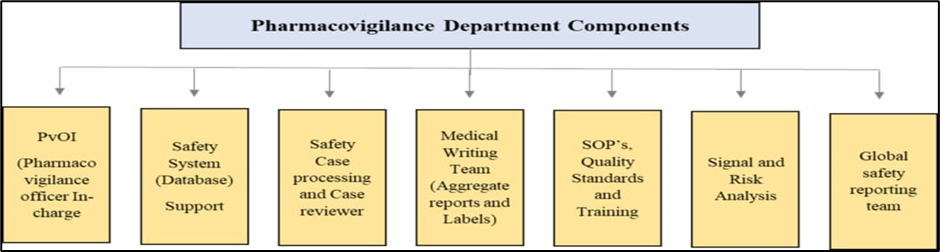
Figure 1: Component of Pharmacovigilance
trials are to be reported by the investigator to the sponsor and to the Ethics Committee within 24 hours and 7 days respectively. These should be reported by the sponsor to the licensing authority within 14 days of occurrence.[2]n Pharmacovigilance in India started from 1986. A formal Adverse Drug Reactions (ADR monitoring system was initiated with 12 regional Centres, each covering a population of 50 million. However, no noteworthy growth was made. Afterward in 1997, India joined the World Health Organization (WHO) and Adverse Drug Reaction (ADR) scrutinizing program based at Uppsala, Sweden but got fail.[3] Hence, after 2005 WHO supported and World Bank-funded National Pharmacovigilance Programme ((NPPV) of India was made operational.[3]
Concept of pharmacovigilance:
WHO defines the Pharmacovigilance (PV) as the pharmacological science relating to the detection, evaluation, understanding and prevention of adverse effects, particularly long term and short-term side effects of medicine. Pharmacovigilance refers to the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects and other drug- related safety problems. Related to this general definition, the underlying objectives of pharmacovigilance are to prevent harm from adverse reactions in humans that arise from the use of health products within or outside the terms of marketing authorization and in relation to the life cycle of these health products.[4][5] The main goal of pharmacovigilance is thus to promote the safe and effective use of health products, in particular by providing timely information about the safety of health products to patients, health-care professionals, and the public. Pharmacovigilance is therefore an activity contributing to the protection of patients and maintaining public health.[4][5]
1.It involves exhibiting the efficacy of drugs by monitoring their adverse effect profile for many years from the lab to the pharmacy.
2.tracking any drastic effects of drugs improving public health and safety in relation to the use of medicines.
3.encouraging the safe rational and cost-effective use of drugs.
4.promoting understanding, education and clinical training in pharmacovigilance.
5.effective communication to the generic public. In addition, providing information to consumers, practitioners and regulators on the effective use of drugs along with designing programs and procedures for collecting and analysing reports from patients.[4][5]
Components of pharmacovigilance system
Based upon the aim and scope of pharmacovigilance, certain elements and capabilities are very essential to a fully functioning pharmacovigilance system as shown in Figure 1. These include as follow [6]:
• A qualified person for pharmacovigilance –Officer In-charge (PvOI) (India)
• Safety system (database) support
• Safety case processing and review
• Medical writing and aggregate reporting
• A sound quality management system including standard operating procedures (SOPs)
• Quality standards, metrics, and training
• Signal detection and risk analysis
• Global safety reporting
Pharmacovigilance program of India
Pharmacovigilance Programme of India (PvPI) is a highly specialized organization of medical science dealing with activities related to detection, assessment, understanding, prevention and control of adverse effects or any other possible drug related problems. An Indian government agency called the Pharmacovigilance Programme of India (PvPI) detects and addresses issues related to medication safety. Receiving reports of adverse medication occurrences and taking appropriate steps to address issues are among its activities. The initiative was started in July 2010 by the Central Drugs Standard Control Organization, with the All India Institute of Medical Sciences in New Delhi serving as the National Coordination Centre. On April 15, 2011, it moved to the Indian Pharmacopoeia Commission in Ghaziabad. After the Thalidomide scandal in the 1960s, pharmacovigilance programs were established in several developed nations. In the 1980s, India established its program. The Central Drugs Standard Control Organization created the current Pharmacovigilance Program of India in 2010 after this broad idea of drug safety monitoring underwent several iterations. The initiative is currently well-integrated with government laws, a research facility run by the Indian Pharmacopoeia Commission, and a regulator acting as a leader. There were 250 centers in India as of 2018 that could handle reports of severe adverse reactions. Training physicians and hospitals to report adverse medication reactions when patients experience them is one of the organization's challenges. Although the Pharmacovigilance Program generates these reports, any clinic should ideally be the source. The goal of the Pharmacovigilance Program is to promote a social norm and culture that encourages reporting drug issues. As a collaborating center, the Pharmacovigilance Programme assists the WHO in developing international policy for other countries to manage their own drug safety programs. Whi le the United States and Europe have pharmacovigilance systems which are developed well in some ways, the Indian programme has more and specialized expertise to apply for the unique circumstances of India. The program's ability to identify side effects in Indian patients using carbamazepine was one of its achievements. People from South Asia are more prone to have issues when using this medicine because of their different genetic makeup, even if it is safer for native Europeans. The Pharmacovigilance Programme was successful in identifying this issue, which other nations would not have been able to do. The Pharmacovigilance Program's creation increased India's appeal as a global location for clinical trial research for multinational businesses. International researchers conducting trials in India must have a thorough understanding of the Caliber of India's pharmacovigilance program. The program works with the World Health Organization on safe pharmaceutical projects both domestically in India and abroad.[7][8] The aims of this program were multiplier to produce knowledge on ADR in the community of Indians; to increase understanding between medical services practitioners of the importance for PV; to actively track the profit risks of medicinal products; to establish unbiased, impartial, evidence-based guidelines on the protection of medicinal products; to urge regulators to pursue safety-related judgments; Convey results to all relevant investor and set up a National Centre of Excellence under national wide guidelines for drug safety.[2][8]
The structure of PvPI consists of the following two committees.[9]
1. Steering Committee
The Steering Committee consists of 10 members with Drug Controller General of India (DCGI), CDSCO, as ex-officio chairman and officer in charge along with other additional members. This committee is responsible for supervising and giving directions to PvPI.
2. Working Group
The Working Group consists of 11 members with secretary-cum-scientific director and Indian Pharmacopoeia Commission as the chairperson. This committee provides technical support to the programme and serves as a,
(a) Signal review panel
(b) Core training panel
(c) Quality review panel.
The objectives of PvPI are given below [10]:
1. To create an international system for patient safety reporting
2. To identify and analyse new signals (ADR) from reported cases
3. To monitor the benefit-risk ratio from marketed medications list
4. To generate evidence-based information on safety of medicines
5. To support agencies like CDSCO in formulating safety related regulatory decisions for medicines
6. To communicate safety information on use of medicines to various stakeholders to minimize risk
7. To create a National Centre of Excellence at par with global drug safety monitoring
8. To collaborate with National Centres for exchange of information and management of data
9. To provide training and support to other National Pharmacovigilance Centres.
Introduction To Clinical Research
The importance of drug trials in promoting health services cannot be over emphasized. New drugs and therapies can improve the quality and lifespan of patients. While it is imperative that the number of clinical trials increase, the Government is also trying to ensure that the rights and safety of the subjects are protected and the quality of the trials performed in India improve to international standards. The regulatory guidelines in terms of serious adverse events (SAEs) reporting, informed consent, compensation in case of injury or death in clinical trials have been recently modified. It is essential that now all clinical trials conducted in India should as per the international conference of Harmonization-Good Clinical Practices Guidelines (ICH- GCP) for clinical trials and follow the recently amended Schedule Y of the Drugs and Cosmetics Act.[11][12]
Clinical Trials
A clinical trial is a research study that tests a new medical treatment or a new way of using an existing treatment to see if it will be a better way to prevent and screen for diagnose or treat a disease. For any new drug to enter in clinical trial, it must pass preclinical studies. Preclinical studies involve in vitro (i.e. test-tube or Laboratory) studies and trials on animal populations. Wide range of dosages of the study drug is given to animal subjects or to an in-vitro substrate in order to obtain preliminary efficacy, toxicity and pharmacokinetic information. It is important for anyone preparing a trial of a new therapy in humans that the specific aims, problems and risks or benefits of a particular therapy be thoroughly considered and that the chosen options be scientifically sound and ethically justified.[11][12]
Phases of trials
1.Phase I or clinical pharmacology study: Initial safety trials on a new medicine. An attempt is made to establish the dose range tolerated by volunteers for single and for multiple doses, pharmacodynamic effects, pharmacokinetics and nature and intensity of adverse reactions (s).
Normally, a small group of 20-100 healthy volunteers will be recruited. These trials are often conducted in a clinical trial clinic, where the subject can be observed by full-time staff. Investigators involved in Phase I trials should be trained in clinical pharmacology and should also be provided with necessary facilities which would help them in closely observing and monitoring the subjects.[12]
2.Phase II or exploratory trials
Phase IIa: Pilot clinical trials to evaluate efficacy and safety in selected populations of patients with the disease or condition to be treated, diagnosed, or prevented. Objectives may focus on dose-response, type of patient, frequency of dosing, or numerous other characteristics of safety and efficacy.
Phase IIb: This trial is to evaluate efficacy (and safety) in patients with the disease or condition to be treated, diagnosed, or prevented. These clinical trials usually represent the most rigorous demonstration of a medicine’s efficacy. Sometimes referred to as pivotal trials. Phase II trials are performed on larger groups (100-300 individuals) and are designed to assess how well the drug works, as well as to continue Phase I safety assessments in a larger group of volunteers and patients. [12]
3.Phase III or confirmatory trials
Purpose is to obtain sufficient evidence about the efficacy and safety of the drug in a larger number of patients, generally in comparison with a standard drug and/or a placebo as appropriate. This also helps in determining the dose-response relationship and its use in wider population in different stages of disease. In this phase, the group is between 1000-3000 subjects.[12]
4.Phase IV trials or post-marketing phase
Studies or trials conducted after a medicine is marketed to provide additional details about the medicine’s efficacy or safety profile. Different formulations, dosages, durations of treatment, medicine interactions, and other medicine comparisons may be evaluated. New age groups, races, and other types of patients can be studied. Detection and definition of previously unknown or inadequately quantified adverse reactions and related risk factors are an important aspect of many Phase IV studies. If a marketed medicine is to be evaluated for another (i.e., new) indication, then those clinical trials are considered Phase II clinical trials. The term post-marketing surveillance is frequently used to describe those clinical studies in Phase IV that are primarily observational or non- experimental in nature, to distinguish them from well controlled Phase IV clinical trials or marketing studies.[12]
Drug Controller General of India (DCGI)
Drug Controller General of India (DCGI) is the full name of the organization. DGCI is in charge of the department known as the Central Drugs Standard Control Organization (CDSCO). The Indian Government is in charge of the department. Licenses for particular drug classes in India must be approved by the DCGI. Drug quality requirements and standards are also outlined in the DCGI. In India, it has to do with producing, distributing, importing, and selling medications. With the Drugs and Cosmetics Act, the DCGI seeks to standardize enforcement.[13]
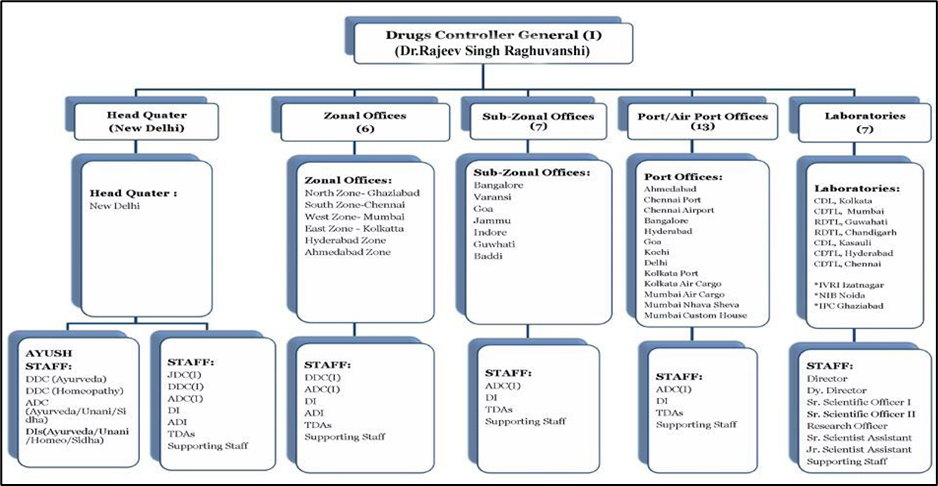
Figure 1: Structure of DCGI [13]
The following are the major functions of DCGI [13]:
Central Drugs Standard Control Organization (CDSCO)
The Central Drugs Standard Control Organization (CDSCO) is the Central Drug Authority for discharging functions assigned to the Central Government under the Drugs and Cosmetics Act. CDSCO has six zonal offices, four sub-zonal offices, 13 port offices and seven laboratories under its control. [14] The following are the major functions of CDSCO [14]: Regulatory control over the import of drugs, approval of new drugs and clinical trials, meetings of Drugs Consultative Committee (DCC) and Drugs Technical Advisory Board (DTAB), approval of certain licenses as Central License Approving Authority is exercised by the CDSCO headquarters.
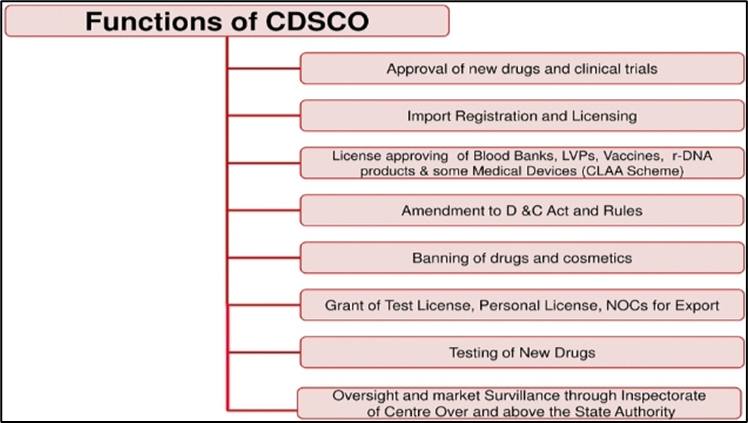
Figure 2: functions of CDSCO [14]
According to the Drug and Cosmetics Act, the regulation of the manufacture, sale, and distribution of drugs is primarily the concern of the state authorities, while the Central Authorities are responsible for the approval of new drugs, clinical trials in the country, laying down the standards for drugs.
Types of regulatory applications:
There are three IND types:
The goals of the NDA are to provide enough information to permit FDA reviewer to reach the following key decisions:
The documentation required in an NDA is supposed to tell the drug’s whole story, including what happened during the clinical tests, what the ingredients of the drug are, the results of the animal studies, how the drug behaves in the body, and how it is manufactured, processed and packaged.[16]
Generic drug applications are termed “abbreviated” because they are generally not required to include preclinical (animal) and clinical (human) data. Instead, generic applicants must scientifically demonstrate that their product performs in the same manner as the innovator drug.
The ANDA must demonstrate that:
Ich good clinical practice
Good Clinical Practice (GCP) training is a basic need for anyone conducting clinical research. GCP is standard and set of rules that apply to all research. GCP is a set of globally accepted standards for scientific and ethical quality that need to be followed at every turn during clinical research. The most promising therapies are advanced into clinical trials after laboratory testing and animal research. GCP or Good Clinical Practice refers to an international quality Standard provided by the ICH for the purpose of regulating clinical Trials that involve human subjects. [18]
The objective of Good Clinical Practice (GCP) is to ensure the safety, rights, and well-being of scientific patients while maintaining the credibility and integrity of data collected in clinical studies
New drug clinical trial rule 2019
New Drugs and Clinical trial rule 2019 are regulated on 19 March 2019. Their objective is to implement a dependable, comprehensible, and uniform methodology for clinical trials. With these rules, Indians will perhaps have faster access to novel medications and clinical trials. The new drug and clinical trial rules 2019, rule 97 includes 122DAA in the drugs and cosmetic rules 1945. It includes the disregard for specific regulations for novel medications and novel drugs under study for use in humans. For new and experimental medications intended for human use, Part XA and Schedule Y will not apply.[20]
Objective NDCT [20][21]:
The new rules include a time-bound evaluation of applications and give additional flexibility to researchers. They are intended to promote clinical research and ensure predictability and accountability in the regulatory process.
Scope of NDCT [20][21]:
Process of clinical trial application
A Clinical Trial Application provides comprehensive information about the investigational medicinal product(s) and planned trial, enabling regulatory authorities to assess the acceptability of conducting the study. Health authorities’ assessment covers the investigational medicinal product properties, the benefit/risk ratio of the study, the quality of the information provided to the trial subjects, and the suitability of the clinical sites and investigators.[22]
Adverse drug reaction monitoring
The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, of which the World Health Organization (WHO) and the United States Food and Drug Administration (FDA) are members, defines an ADR as "A response to a drug which is noxious and unintended, and which occurs at doses normally used for prophylaxis, diagnosis, or therapy of disease or the modification of physiologic function." An adverse drug event, on the other hand, is defined as: "Any untoward medical occurrence that may present during treatment with a pharmaceutical product, but which does not
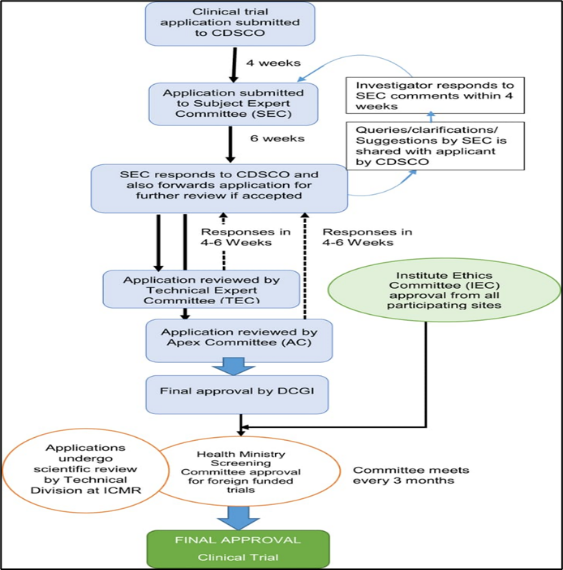
Figure 3: Process of clinical trials
necessarily have a causal relationship with this treatment." [23] ADR monitoring is spelled out as the practice of continuously monitoring the undesirable effects caused using any Drug. Pharmacovigilance plays an imperative impersonation in monitoring ADRs. It Is inherent for pharmaceutical regulators to screen their pharmaceutical products in the market and record if any suspected adverse reactions are identified. The introduction of this monitoring procedure intends at warranting that patients receive safe and beneficial medicinal products. If any of the adverse events are not stated it may result in noxious and serious effects of remedial products. Thus, properly conducting ADR monitoring programmes will Help to reduce the harmful effects of therapeutic products. [24]
Table 1: List of ADR Monitoring centre [25]
|
List of 760 AMCs under PvPI |
||||
|
Sr. No |
AMC location |
Total No. Of centres |
Total No. Of government centre |
Total No. Of non-government centre |
|
|
Andhra Pradesh |
42 |
12 |
30 |
|
|
Arunachal Pradesh |
2 |
2 |
0 |
|
|
Assam |
12 |
10 |
2 |
|
|
Bihar |
12 |
6 |
6 |
|
|
Chhattisgarh |
9 |
5 |
4 |
|
|
Goa |
1 |
1 |
0 |
|
|
Gujrat |
42 |
10 |
32 |
|
|
Haryana |
35 |
5 |
30 |
|
|
Himachal pradesh |
9 |
7 |
2 |
|
|
Jammu and Kashmir |
10 |
9 |
1 |
|
|
Jharkhand |
71 |
21 |
50 |
|
|
Keral |
50 |
10 |
40 |
|
|
Madhya pradesh |
21 |
11 |
10 |
|
|
Maharashtra |
64 |
20 |
44 |
|
|
Manipur |
3 |
2 |
1 |
|
|
Meghalaya |
1 |
1 |
0 |
|
|
Mizoram |
2 |
2 |
0 |
|
|
Odisha |
15 |
9 |
6 |
|
|
Punjab |
29 |
4 |
25 |
|
|
Rajasthan |
36 |
14 |
22 |
|
|
Sikkim |
2 |
0 |
2 |
|
|
Tamil Nadu |
66 |
28 |
38 |
|
|
Telangana |
56 |
8 |
48 |
|
|
Tripura |
2 |
1 |
1 |
|
|
Uttar pradesh |
76 |
33 |
43 |
|
|
Uttarakhand |
8 |
4 |
4 |
|
|
West Bengal |
29 |
15 |
14 |
|
|
Andhra Pradesh |
1 |
1 |
0 |
|
|
Chhattisgarh |
1 |
1 |
0 |
|
|
Ladakh |
1 |
1 |
0 |
|
|
New Delhi |
33 |
11 |
22 |
|
|
Pondicherry |
7 |
3 |
4 |
|
|
Diu dam and &haveli |
1 |
1 |
1 |
Function of AMCs [26]:
Design and conduct of observational studies
Carefully designed and conducted pharmaco-epidemiological studies, specifically observational (non-interventional, non-experimental) studies, are important tools in pharmacovigilance. In observational studies, the investigator "observes and evaluates results of ongoing medical care without 'controlling the therapy beyond normal medical practice.[27] Observational studies, crucial in pharmacovigilance, involve monitoring medical care without altering standard practices. Before starting such studies, a finalized protocol should be prepared, consulting experts like pharmacovigilance professionals and biostatisticians. Regulatory authorities should review the protocol and early termination conditions. Study protocols must outline objectives, methods, and analysis plans. Upon completion, a detailed report should be submitted. Adherence to good epidemiological practices, international guidelines, and local laws is essential. Professional conduct and confidentiality, along with data protection laws, must be upheld throughout the study.[28]
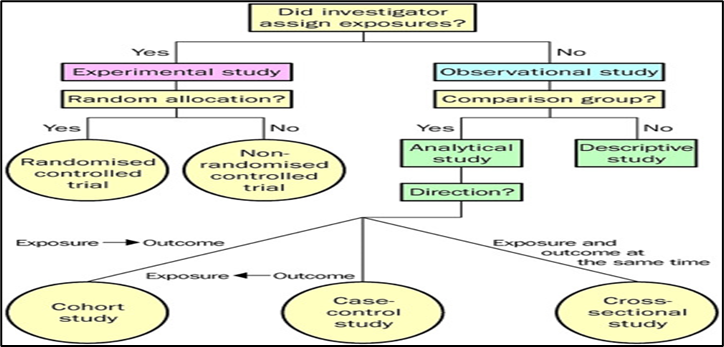
Figure 5: Classification of observational studies
Classification of observational studies:
Adverse Drug Reaction Monitoring Form
A "Drug Monitoring Form" is used to collect and document data regarding the safety, efficacy, and adverse effects of a drug. In the context of Indian Pharmacopoeia (IP) or the Indian regulatory system, drug monitoring is particularly associated with pharmacovigilance. Below is an outline of a typical drug monitoring form (IP). Information provided in this form is handled in strict confidence. The causality assessment is carried out at AMCs by using WHO-UMC scale. The analyzed forms are forwarded to the NCC-PvPI through ADR database. Finally the data is analyzed and forwarded to the Global Pharmacovigilance Database managed by WHO Uppsala Monitoring Centre in Sweden.The reports are periodically reviewed by the NCC-PvPI. The information generated on the basis of these reports helps in continuous assessment of the benefit-risk ratio of medicines.[29]
![Figure 4 Naranjo scale [30].png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20241212170736-1.png)
Figure 4: Naranjo scale [30]
![Naranjo scale [30].png](https://www.ijpsjournal.com/uploads/createUrl/createUrl-20241212170736-0.png)
Following is the suspected adverse reaction monitoring form as per indian pharmacopoeia.
Assessment of ADR by naranjo scale
The Naranjo Adverse Drug Reaction (ADR) Probability Scale is a tool used to determine the likelihood that an adverse event is related to a drug rather than other factors. It provides a structured way to assess causality, commonly used in clinical settings and pharmacovigilance.
The scale consists of 10 questions, each with a possible score of -1, 0, +1, or +2 depending on the answer. The total score helps categorize the adverse event:
? 9: Definite ADR
5-8: Probable ADR
1-4: Possible ADR
? 0: Doubtful ADR
Table 2: Naranjo Algorithm - ADR Probability Scale [31]
|
Score |
Interpretation of Scores |
|
Total Score |
Definite. The reaction (1) followed a reasonable temporal sequence after a drug or in which a toxic drug level had been established in body fluids or tissues, (2) followed a recognized response to the suspected drug, and (3) was confirmed by improvement on withdrawing the drug and reappeared on reexposure. |
|
Total Score |
Probable. The reaction (1) followed a reasonable temporal sequence after a drug, (2) followed a recognized response to the suspected drug, (3) was confirmed by withdrawal but not by exposure to the drug, and (4) could not be reasonably explained by the known characteristics of the patient’s clinical state. |
|
Total Score |
Possible. The reaction (1) followed a temporal sequence after a drug, (2) possibly followed a recognized pattern to the suspected drug, and (3) could be explained by characteristics of the patient’s disease. |
|
Total Score |
Doubtful. The reaction was likely related to factors other than a drug. |
CONCLUSION:
Pharmacovigilance plays a crucial role in ensuring medication safety and protecting public health. It is noticed that Effective pharmacovigilance systems can significantly reduce adverse drug reactions and improve patient outcomes. Also, Continuous monitoring and evaluation of pharmacovigilance practices are essential for optimizing medication safety.[32] As per study, it is concluded that Healthcare professionals should prioritize pharmacovigilance in their daily practice and also made Patients actively involved in medication safety through education and awareness. In advance, Further studies are needed to explore the application of artificial intelligence in pharmacovigilance the Research should focus on developing novel pharmacovigilance metrics and evaluation tools for better use of pharmacovigilance in upcoming generation.[3][33]
REFERENCES


Pratiksha Salunkhe*, Sunil Yadav, Harshada Jadhav, Dr. Shoheb Shaikh, Pharmacovigilance and the Process of ADR reporting and Monitoring in India, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 1585-1599. https://doi.org/10.5281/zenodo.14412861
 10.5281/zenodo.14412861
10.5281/zenodo.14412861
