1Assistant Professor, Department of Quality Assurance, Tathya Pharmacy College, Chikhli.
2Principal, Department of Pharmaceutics, Tathya Pharmacy College, Chikhli.
Impurity profiling plays a crucial role in the pharmaceutical industry to ensure the safety, efficacy, and quality of drug products. This review provides an overview of impurity profiling methodologies, focusing on pharmaceutical drug candidates. It begins with an introduction to impurities, including their classification and sources. Moreover, the importance of impurity thresholds and regulatory guidelines established by organizations like the International Council for Harmonisation (ICH) and the United States Pharmacopeia (USP) is highlighted. Furthermore, strategies for impurity control during drug development stages are examined, encompassing synthesis, formulation, and manufacturing processes. The significance of impurity profiling in ensuring drug product safety, stability, and regulatory compliance is emphasized throughout. This comprehensive overview aims to assist researchers, pharmaceutical scientists, and regulatory professionals in understanding the importance of impurity profiling and implementing effective strategies for impurity control in pharmaceutical drug development.
Consumer’s protection depends on a products safety, characteristics, purity of the components. All these are regulated by The U.S. Food and Drug Administration (FDA). Small amount of impurity can change the efficacy, toxicity of any pharmaceutical compounds. International Conference on Harmonization said that impurities are unwanted chemicals that remain with the Active Pharmaceutical Ingredients (APIs) or develop during formulation or develop upon ageing of both APIs and formulated APIs. As safety and quality of pharmaceutical products can be affected by the impurities present in the Active Pharmaceutical Ingredients (APIs) the impurity profile study of the API to be used in the manufacturing of drug substance. Thus, impurity profiling like identification, Isolation & characterization are done and their threshold values comply with the limits set and specified by official bodies. Issue related to impurities? addressing must be the same for each and every sectors and there must be a unified system to ensure it. International Conference on Harmonization (ICH) has published guidelines for validation methods for analysis of impurities in new drug products, new drug substances, residual solvents & microbiological impurities [4-6] for registration of pharmaceuticals. ICH defines impurities as ?substance in the API itself.? For pharmaceutical products, impurities are defined as ?substances in the product that are not the API itself or excipients used to manufacture it.? i.e. impurities, are unwanted chemicals that remain within the formulation or API in small amounts which can influence QSE, thereby causing serious health Hazards.
Regulatory Guidelines on impurity
International Conference on Harmonization guidance of Technical Requirements for Registration of Pharmaceuticals for Human Use is inscribed by The United States Food and Drug Administration (FDA).
The FDA has the assigned responsibility of ensuring the safety and efficacy of drugs. The various regulatory guidelines [2] regarding impurities are as follows:
Qualification of Impurities
The impurity profile of drug substance may vary for processes like scale-up changes, synthetic route change and changes made to key intermediates. New Molecular Entities (NMEs) limits are classified and restricted by the ICH. Studies are needed to be done to ensure that the impurity limits does not exceed beyond the range given in the table. Qualification process helps to acquire and evaluate data that establishes the biological safety of an individual impurity.

Table:- 1 Threshold Limits
Compound investigated in drug discovery leads to a significant analytical challenge for the characterization, quantization, and detection of the compounds. Here, in following Figure, we have summarized all classes of impurities.
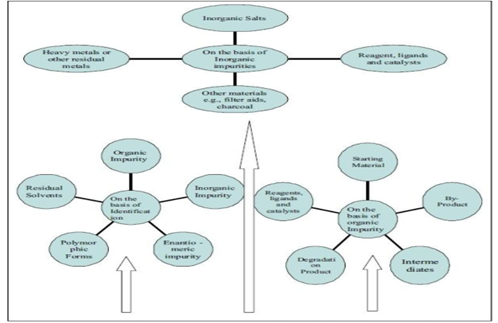
Impurities in pharmaceutical compounds are mainly formed through the synthesis process as the product is contaminated by raw materials, solvents, intermediates and by-products. A general idea of these impurities is given below.

Table:- 2 Sources of impurity
These types of impurities form during the manufacturing process or during storage of the drug substance. The sub- types of these impurities are given below.
During multistep synthesis process there are high chances of impurities formed as by products, intermediates are produced. So, special care is needed. It results in unreacted starting material in the final product.
Example:
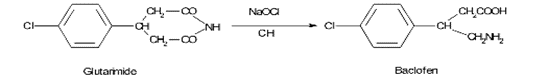
In the synthesis of Baclofen, the last step carried out with ?-(p-chlorophenyl) gutarimide, which on reaction with NaOH/sodium hypochlorite solution at room temperature yields a potential impurity p-chlorophenyl glutaric acid, which has to be evaluated.
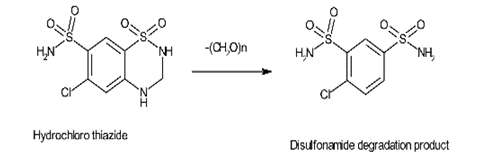
Product degradation happens during the synthetic process, storage, formulation of dosage form and aging. Authoritative examples for impurities from degradation products are penicillin and cephalosporin. Another degradation pathway is shown in Hydrochlorothiazide through which it degrades to the disulfonamide in its synthesis.
In organic chemistry 100% pure product is not generally formed as there is always a chance of having by-products. By products can be formed through variety of side reactions, such as incomplete reaction, rearrangement, dimerization, over reaction, isomerization or unwanted reactions between starting materials. For example diacetylated paracetamol may forms as a by-product In the case of paracetamol production.
Inorganic impurities are also obtained from the manufacturing processes which are used in bulk drug formulation. They are normally known and identified.
Single enantiomeric form of a chiral drug provides greater chemical entity. It also helps to provide better therapeutic Index. Conversely, the pharmacokinetic profile of ofloxacin (R-isomeric form) and levofloxacin (S-isomeric form) are similar, suggesting the lack of advantages of single isomer.
These impurities are pretty rare. Proper care during the manufacturing process avoids the chance of these kinds of impurities.
Water is essential during manufacturing process and it is the main source of heavy metals, like Ar, Cd, Cr, Na, Mg, Mn, etc. These can be avoided by the use of demineralization plant, reverse osmosis technique that produces mineral free water.
The filters or filtering aids are routinely used in the bulk drugs manufacturing plants and sometimes activated carbon is also used which acts as a source of impurity. For that reason regular monitoring of fibres and black particles are needed to avoid the contamination.
They are potentially undesirable substances which either hazardous to human health or modify the properties of certain compounds. The residual solvents also affect physicochemical properties like crystalline of bulk drug, which affect the dissolution properties, colour changes in finished products. ICH classified these substances in to four types.
a. Class I solvents
These solvents are either avoided or restricted to a limit in the manufacture of excipients and drug substances because of their unacceptable toxicity or their deleterious effects. These are generally carcinogens.
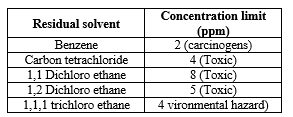
Table:- 3 Class I Residual Solvents
As Class II solvents are inherently toxic, their usage should be limited in pharmaceutical Industry. These are generally Non-genotoxic, animal carcinogens and possible neurotoxicants.
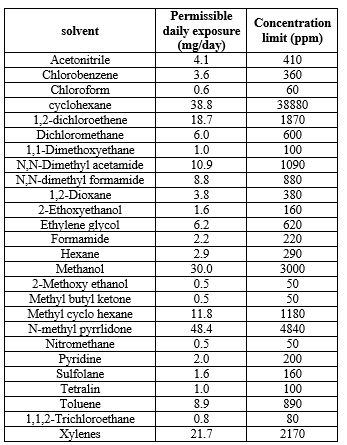
Table:- 4 Class II Solvents with Their Permissible Daily Exposure Limits
As they are less toxic and possess lower risk to human health than class I or class II solvents, they do not have any serious health hazard. According to several data‘s, long term toxicity is generally not reported.
Formulation-related impurities
Drug substance varies with conditions that lead to its degradation or other chemical reactions. Solutions and suspensions are prone to degradation due to hydrolysis. Water used in formulation contribute to not only its impurity but also provide stimulation for process like hydrolysis and catalysis.
The formulation related impurities can be classified as follows:
The primary environmental factors that can reduce stability can be sub classified
Dosage form related
Metabolite impurities
By products formed by drugs after instigation in body are generally known as Metabolite impurities. Metabolite impurities can be formed during metabolism as the API and drug product in the body is exposed to various enzymes.
Examples are asenapine N-oxide, asenapine desmethyl, and ciprofoxacin ethyl diamino impurity, which are formed as process impurities, but are also metabolites of the same process.
Analytical method development
Meaningful and reliable analytical data is needed to produce new drug various stages of the development. Sample set selection for analytical method development Screening of Chromatographic conditions and Phases, typically using the linear solvent- strength model of gradient elution. Optimization of the method to fine-tune parameters related to ruggedness and robustness
The impurities can be identified predominantly by following methods:
Table:- 5 Achiral method development process
Remedies
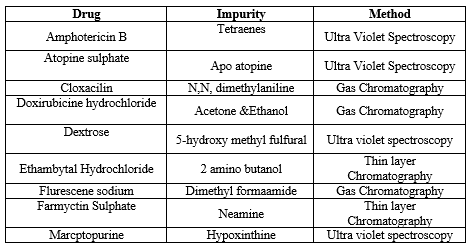
Table:- 6 Current marketed formulation which contain impurity
Applications
Numerous applications have hunted for the areas of drug designing, in monitoring quality, stability, and safety of pharmaceutical compounds. The applications include alkaloids, amines, analgesics, anticonvulsants, antidepressant, tranquilizers, antineoplastic agents, macromolecules, steroids etc.

Table:- 7 Goals of impurity investigations
CONCLUSION:
In conclusion, impurity profiling serves as a crucial aspect of pharmaceutical drug development, enabling the thorough characterization of potential contaminants and degradation products within drug candidates. Through comprehensive analysis, we have identified and quantified various impurities present in the drug substance, elucidating their origins and potential impact on product quality, safety, and efficacy. This information is invaluable for regulatory compliance and ensuring the production of high-quality pharmaceuticals. Moving forward, continued monitoring and optimization of impurity profiles will be essential to mitigate risks and enhance the overall quality of the drug product.
REFERENCES:


Mansi Rana, Vikram Pandya, Overview On Impurity Profiling For Pharmaceutical Drug Candidates, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 4, 586-593. https://doi.org/10.5281/zenodo.10955539
 10.5281/zenodo.10955539
10.5281/zenodo.10955539
