Department of Pharmaceutics, Shree Dev Bhoomi Institute of Education Science & Technology, Pondha Rd. Majhaun, Uttarakhand
The development and assessment of non-effervescent tablets are essential for ensuring the controlled release of active ingredients such as Pregabalin. Non-effervescent tablets are specifically designed to release medication gradually and consistently over an extended period, which is particularly advantageous for drugs like Pregabalin utilizing HPMC K100M and Carbopol 971P as release-retarding polymers. The objective of this study was to create and assess controlled release matrix tablets containing Pregabalin. The main purpose of this study was to develop gastroretentive tablets with floating and swelling properties for once-daily administration of pregabalin. The non-effervescent floating and swelling tablets were prepared using wet granulation and compaction, which are widely used and easily accessible. The amount of hydroxypropyl methylcellulose and crospovidone were found to be critical factors affecting in vitro dissolution and floating properties of the prepared tablets. The rationale emphasizes the limitations of immediate-release Pregabalin formulations, such as the need for multiple daily doses and potential fluctuations in drug levels, which might lead to inadequate pain control and reduced patient adherence. It underscores the necessity for a formulation that offers sustained pain relief with less frequent dosing. Enhancing Patient Compliance.: By reducing dosing frequency through controlled release, the research aims to enhance patient compliance and convenience.
The primary method for administering drugs for systemic effects is oral drug administration with 90% of all drugs used to produce its result [1]. Among various oral route of drug administration, sustained release dosage forms are designed to enhance the pharmaceutical activity of the drug, reduce dosage frequency and side effects, and improve patient convenience. Gastro retentive drug delivery systems are designed to be retained in the stomach for extended periods, allowing for sustained and prolonged drug input to the upper part of the gastrointestinal tract [2]. These systems are particularly useful for drugs acting locally in the stomach, having narrow absorption window, unstable in the intestinal or colonic environments, or having low solubility at high pH values. Techniques for forming successful gastro retentive drug delivery systems include floating dosage forms, low density systems, raft systems, bio adhesive or muco-adhesive systems, high density systems, super-porous hydrogel, and magnetic systems. These systems offer advantages such as sustained blood levels, attenuation of adverse effects, and improved patient compliance. Active pharmaceutical ingredients (APIs) are chemical-based compounds produced mainly in the USA, Europe, China, and India. Modern medicines consist of two main components: chemically and biologically active components [3]. Excipients in dosage forms play a significant role in the final product, including manufacturing, stability, dose uniformity, effective delivery, and organoleptic properties [4]. Pharmaceutical excipients are typically included in dosage forms in larger quantities than the API, making up to 90% of the total mass/volume of medicinal products [5].
AIM
OBJECTIVE
Solid dosage forms are popular among others because of their low cost, ease in administration, self-medication, accurate dosage, pain avoidance and good patient compliance. Tablets and capsules are most popular solid dosage forms. Pregabalin is rapidly absorbed with peak plasma concentration occurring at around 1 hour of administration. since pregabalin has narrow therapeutic absorption window, design of once daily sustained release tablet that release drug at controlled rate and also retains in upper part of GIT for a longer period of time would be relevant. This dosage form cause reduction in frequency of dose hence enhance patient compliance and reduce adverse effect in comparison to immediate release. Plasma concentration of pregbalin can be maintained for long period of time and increased bio-avability of drug can remain as required.
Addressing Treatment Limitations:
The rationale emphasizes the limitations of immediate-release Pregabalin formulations, such as the need for multiple daily doses and potential fluctuations in drug levels, which might lead to inadequate pain control and reduced patient adherence. It underscores the necessity for a formulation that offers sustained pain relief with less frequent dosing. Enhancing Patient Compliance.: By reducing dosing frequency through controlled release, the research aims to enhance patient compliance and convenience. The rationale might discuss how this improved dosing regimen could positively impact patient adherence to the treatment plan, leading to better pain management outcomes.
Stable Drug Levels:
Highlighting the importance of maintaining consistent therapeutic levels of Pregabalin in the body, the rationale stresses the significance of the controlled-release mechanism in achieving steady-state drug concentrations. This stability is crucial for sustained pain relief and minimizing fluctuations that may lead to breakthrough pain.
Optimizing Pharmacokinetics:
The research aims to optimize Pregabalin's pharmacokinetics by developing a formulation that releases the drug gradually over an extended period. This sustained- release profile seeks to achieve a balance between efficacy and safety, ensuring a controlled and predictable drug delivery system.
Improving Quality of Life:
Emphasizing the impact on patients' quality of life, the rationale discusses how a more convenient dosing schedule and consistent pain relief can positively influence daily functioning, reduce the burden of managing chronic pain, and potentially improve overall well-being.
Competitive Advantage:
Exploring the competitive landscape, the rationale might discuss the potential market advantage of introducing Lyrica CR. It could emphasize the potential for differentiation in the market by offering a unique formulation that addresses the drawbacks of existing immediate-release versions of Pregabalin.
Clinical and Economic Benefits:
In addition to clinical advantages, the rationale might touch upon potential economic benefits, such as reduced healthcare costs associated with improved patient adherence, fewer hospital visits due to better pain control, and potentially lower overall healthcare utilization.
Advantages:
DRUG PROFILE:
Pregabalin is a calcium channel blocker that inhibits calcium influx into nerve cells, reducing neuronal hyper-excitability and contributing to its analgesic effects [6]. It is used for treating seizure disorders, fibromyalgia, and neuropathic pain. The pharmacological activity of pregabalin is known for its stereo isomeric structure, (S)-enantiomer, which has gained importance in the pharmaceutical industry [7]. Neuropathy pain (NP) is characterized by sensory pathway dysfunction and can be of high intensity and long duration, impacting mood, personality, and social relationships. Pregabalin is effective for both central and peripheral NP and achieves rapid pain reduction. Recently, the US FDA approved once-daily sustained release tablets for post-therpetic neuralgia. However, the product must be taken after an evening meal and a high calorie diet to ensure the same extent of absorption.

Fig 1. Structure of pregabalin
Physical Description
Table 1. Physical Description of pregabalin

The current study aims to develop oral controlled release matrix tablets of Pregabalin, using hydrophilic matrices like carbopol 971P, HPMC K100M and Crospovidone.
MATERIALS AND METHODS
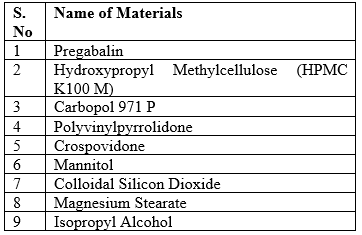
Materials
Methodology:
Solubility
Solubility test was performed as a part of purity test. Solubility of PGB was measured by taking 50 mg of drug in a test tube. 0.1 ml of solvent was added stepwise with shaking. Addition of solvent was continued till the sample was dissolved completely. Solubility was determined in form of the solvent required for solubilisation of the drug powder.[8]
Loss on Drying[9]
Weighing bottle was dried in an oven & allowed to cool to room temperature in a desiccator over silica gel and weighed (W1). 1 g of PGB sample was taken in the dried weighing bottle and weighed again (W2). The bottle containing PGB sample was dried at 105oC for 3 hours. After drying, the weighing bottle was allowed to cool to room temperature in a desiccator over silica gel & weighed (W3). The Loss on Drying of PGB drug was calculated as:
|
Loss on drying % |
= |
W2 – W3 |
x 100 |
|
W2 – W1 |
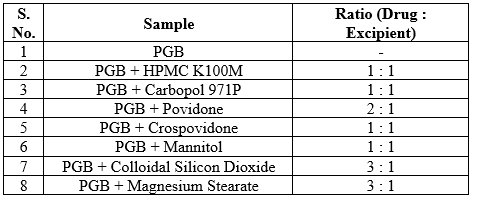
Drug – Excipient Compatibility Study (Isothermal Stress Testing) [11]
Isothermal stress testing was conducted to determine the compatibility of PGB with different excipients. For isothermal stress testing, drug and different excipients were accurately weighed and transferred to a 25 ml volumetric flask (n = 3) and mixed on a vortex mixer for 2 min. In each of the flasks, 10% (w/w) water was added and the drug–excipients blend was further mixed on a vortex mixer. Each flask was closed with stopper, wrapped with parafilm and stored at 50oC in a hot air oven. These samples were periodically examined for any unusual color change. After 3 weeks of storage at the above conditions, samples were suitably diluted and quantitatively analyzed using HPLC method for assay of Pregabalin raw material. Similarly, drug–excipients blends without added water were stored in a refrigerator at 8oC which served as controls.
Table 2 : Ratio of Drug and Excipient blend for Isothermal Stress Testing
Pre-Formulation Studies
First the dosage formulation was studied to generate information useful in formulating acceptable, safe, stable. Different parameters like angle of repose, bulk density, true density, compressibility index, Hausner ratio were evaluated. Angle of repose is evaluated from funnel method. Bulk density is evaluated from bulk density apparatus.
In vitro Dissolution Studies
The in-vitro dissolution studies were performed using the USP-II (Paddle) dissolution apparatus (Lab India) at 50 rpm. Dissolution media used was 6.0 pH Methanol buffer maintained at 37±0.5oC. A 5 ml was withdrawn at specific time intervals and same volume of fresh medium was replaced. The withdrawn samples were diluted with pH 6.0, filtered and analyzed on chromatogram using pH 6.0 methanol buffer as a blank. Percentage cumulative drug release was calculated.
Pre-formulation evaluation
Evaluation of Pre-Compression Parameters of Powder Blend
Flow properties and compressibility of powder blends ready for compression were evaluated by determining angle of repose, bulk density, tapped density, Carr’s index and Hausner ratio.
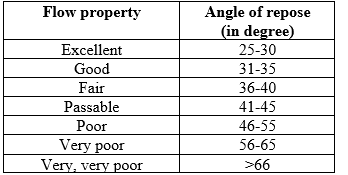
Angle of Repose
Angle of Repose was determined by the fixed funnel method. Angle of repose is the angle formed by the horizontal base and the edge of a cone-like pile of powder. A glass funnel was used with the size of the orifice of 10 mm. The funnel was fixed in place and 10 gram of powder sample was passed through the funnel freely onto the surface. Height (h) and radius (r) of the powder conical pile were measured, and the angle of repose (?) was calculated as follows:
? = tan-1 (h/r)
Table 3: Flow Properties and Corresponding Angles of Repose as per USP [12]
Bulk Density
Bulk density of powder was determined by introducing accurately weighed quantity of powder (10 gram) into a 50 ml graduated measuring cylinder and the bulk volume of the powder was noted.[13] Bulk density of powder was calculated as follows:
Tapped Density
Tapped density of powder was determined by introducing accurately weighed quantity of powder (10 gram) into a 50 ml graduated measuring cylinder and the cylinder was fixed on the tapped density apparatus. The powder was subjected to tapping in the tapped density apparatus until constant volume was obtained.[13] The final volume was noted and tapped density of powder was calculated as follows:
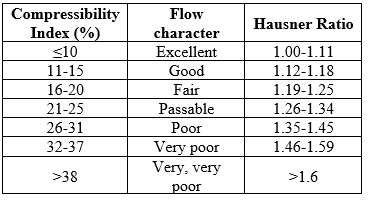
Carr’s Compressibility Index
Compressibility is an indication of ease with which a material can be induced to flow.[14] Carr’s Compressibility Index of the powder was determined as:
Hausner Ratio
Hausner ratio is a measure of the interparticulate friction, which could predict powder flow properties. [15]
Table 4: Scale of Flowability as per USP [12]
Evaluation of Non-effervescent Gastro-retentive Tablets of Pregabalin
Drug Content
Pregabalin content in tablets was determined by comparing with the reference standard of Pregabalin using HPLC method as follows: [10]
Chromatographic Condition:
Standard Solution:
400 mg of Pregabalin standard was dissolved in 100 ml of mobile phase to obtain a solution of concentration 4 mg/ml.
Sample Solution:
Ten tablets were taken, crushed and the crushed powder was mixed. In a 100 ml volumetric flask, a quantity of powder containing about 400 mg of Pregabalin was dispersed with 70 ml of the mobile phase, sonicated for 10 minutes and dilute to 100 ml with the mobile phase.
System Suitability Requirements:
Standard Solution was injected. The test was not valid unless the number of theoretical plates was not less than 2000, tailing factor of the peak of Pregabalin was not more than 2.0 and the relative standard deviation for replicate injections was not more than 2.0 %. Standard Solution and Sample Solutions were injected into the HPLC system. The percentage of Pregabalin in the portion of tablets taken was calculated as:
|
% Drug Content |
= |
Aspl |
x |
Wstd |
x |
50 |
x |
Assay of std in % |
x |
Avg. Wt. |
x |
100 |
|
Astd |
x |
50 |
x |
Wspl |
x |
300 |
|
|
|
|
where,
Astd = Area of Standard
Wstd = Wt. of standard (in mg)
Aspl = Area of Sample
Wspl = Wt. of sample (in mg)
300 = Claim of Pregabalin (mg)
Avg. Wt. = Avg. wt. of tablets (in mg)
Tablet Swelling Ability
The swelling behaviour of the tablets was determined, in triplicate, as per the method described by Gangurde et al. [16] A tablet was weighed (W1) and placed in Type-II dissolution apparatus containing 900 ml of 0.06 N HCl at 37 ± 0.5°C. The medium was stirred continuously at 50 rpm. After 2 hour, 4 hour, 8 hour and 24 hour, the tablets were removed from the dissolution vessel, the excess surface liquid was removed carefully using filter paper and the swollen tablet was then reweighed (W2).[17] The swelling index (SI) was calculated using following formula
|
Swelling Index |
= |
W2 - W1 |
|
W1 |
In vitro Buoyancy Study
The floating behaviour of the tablets was determined visually, in triplicate, according to the floating lag time method described by Rosa et al.[18] A tablet was placed in a 250 ml glass beaker, containing 200 ml of 0.06 N HCl, maintained in a water bath at 37 ± 0.5 ºC. The floating lag time, ‘‘the time between tablet was placed in a glass beaker with HCl and its buoyancy” and total floating duration, ‘‘the time during which tablet remains buoyant”, were recorded. Floating lag time and total floating time was also determined, in triplicate, by placing a tablet in 900 ml of 0.06N HCl in USP type II dissolution apparatus (37 ± 0.5°C, 50 rpm) as described by Gharti et al.[19] The time required for the tablets to rise to the surface and float was determined as floating lag time and the duration of time the table constantly remained on the surface was determined as the total floating time.
Tablet Adhesion Retention Period
The adhesion retention period of the tablets was evaluated, in triplicate, by an in vitro method reported by Nakamura et al.[20] An agar plate (2% w/w) was prepared in 0.1 N HCl. A side of the tablet was wetted with 0.1 N HCl and attached to the centre of agar plate by applying a light force with a fingertip.[21] Five minutes later, the agar plate was attached to a disintegration test apparatus and moved up and down in 0.1 N HCl at 37±2oC. The tablet adhered on the plate was immersed into the solution at the lowest point and got out of the solution at the highest point. The retention period of the tablet on the agar plate was visually observed and noted.
In vitro Drug Release Study
The drug release study of prepared Pregabalin non-effervescent gastro-retentive tablets was performed using USP Type II (Paddle) Dissolution Test Apparatus as per the USFDA Dissolution Methods Database for Pregabalin Extended-Release Tablet. The analysis of drug release from the tablets was performed using HPLC method as per Indian Pharmacopoeia 2022 for Pregabalin Capsules Dissolution. [10]
Dissolution Parameters:

Standard Solution:
33 mg of Pregabalin Standard was dissolved in 100 ml of dissolution medium.
Sample Solution:
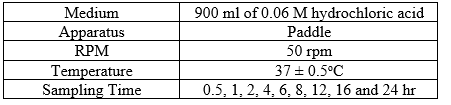
Dissolution parameters were set using 900 ml of 0.06M hydrochloric acid as a medium, one tablet was placed in each dissolution vessel and immediately started the apparatus. At the end of each sampling time points, 20 ml of the medium was withdrawn, filtered and the dissolution vessel was replenished with 20 ml of 0.06M hydrochloric acid (same as volume withdrawn). The filtrate was used as sample.
Chromatographic Condition:
System Suitability Requirements:
Standard Solution was injected. The test was not valid unless the number of theoretical plates was not less than 2000, tailing factor of the peak of Pregabalin was not more than 2.0 and the relative standard deviation for replicate injections was not more than 2.0 %. Standard Solution and Sample Solutions were injected into the HPLC system. The percentage of Pregabalin released was calculated as:
|
% Released |
= |
Aspl |
x |
Wstd |
x |
900 |
x |
Assay of std in % |
x |
100 |
|
Astd |
x |
100 |
x |
1 |
x |
300 |
|
|
where,
Astd = Area of Standard
Wstd = Wt. of standard (in mg)
Aspl = Area of Sample
300 = Claim of Pregabalin (mg)
The time required for 25% of drug release (t25), 50% of drug release (t50) and 80% of drug release (t80) was calculated.
Determination of Difference and Similarity Factors
The in vitro drug release profile of the formulations was compared with the release profile of reference product Lyrica CR (Patent No. US10022447B2) [22] by determining the difference factor (f1) and similarity factor (f2). [23,24] The difference factor (f1) measures the percent error between the two curves over all time points and was calculated as:
where ‘n’ is the number of sampling points, Rt and Tt are the percent dissolved of the reference and test products at each time point ‘t’ respectively.
The similarity factor (f2) is a logarithmic transformation of the sum of squared error of differences between the test Tt and the reference products Rt over all time points. It was calculated as:
The f1 value is 0 when the test and the reference profiles are identical and increases proportionally with the dissimilarity between the two profiles. The f2 value is between 0 and 100. The value is 100 when the test and the reference profiles are identical and approaches zero as the dissimilarity increases.
CONCLUSION
In conclusion, the preparation and evaluation of non-effervescent tablets for controlled release of Pregabalin require precision and expertise. By understanding the nuances of formulation and employing stringent quality control measures, pharmacists can create effective medications that meet the needs of patients. Mastering this process is key to unlocking the full potential of controlled-release drug delivery systems.
REFERENCES

Chandani Rajak , Meenakshi Kandwal, Shivanand Patil , Non-Effervescent Gastro-Retentive Tablets For Controlled Release Of Pregabalin, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 9, 732-742. https://doi.org/10.5281/zenodo.13764897
 10.5281/zenodo.13764897
10.5281/zenodo.13764897
