GRT Institute of Pharmaceutical Education and Research Tiruttani-631209.
Statins are widely prescribed cholesterol-lowering medications that inhibit HMG- CoA reductase. This study employed molecular docking simulations to investigate the binding modes and energies of Atrovastatin and Fluvastatin to various protein targets. This study contributes to the understanding of statin- protein interactions and may aid in the development of novel statin-based therapeutics. The results revealed varying degrees of binding affinity, with Atrovastatin exhibiting stronger binding energies. Analysis of the docking poses and interaction energies provided valuable insights into the structure- activity relationships of these compounds..
Cancer, also known as malignancy, malignant tumor, or neoplasm, occurs when cells grow uncontrollably and invade other tissues. These excess cells can form a tumor. Tumors can be benign or malignant. Benign tumors are non-cancerous, often removable, and rarely return, while malignant tumors are cancerous, capable of spreading to other tissues and organs. This spread is known as metastasis. Cancers are typically named after the organ or cell type where they originate, such as colon cancer or melanoma, which arises from skin melanocytes. Molecular docking is a technique used to predict how a ligand binds to a protein, crucial in drug discovery for optimizing binding affinities. Statins, like Atorvastatin and Fluvastatin, inhibit HMG-CoA reductase to lower cholesterol, with varying efficacy and side effects. This study uses docking simulations to explore their binding modes to protein targets, aiming to optimize statin-based therapeutics. Docking methods include scoring functions, molecular dynamics, and QM/MM models. Advances in medicinal chemistry have shifted from organic chemistry to biotechnology, with high-throughput technologies and bioinformatics leading the way. Statin repurposing is being explored for conditions beyond cholesterol regulation, including anti-cancer effects. Combinatorial chemistry aids in discovering drug leads quickly, and techniques like QSAR and computer-aided drug design (CADD) are critical in optimizing drug properties. ADMET (absorption, distribution, metabolism, excretion, toxicity) is vital for evaluating drug safety. Molecular docking identifies optimal binding sites and interactions between proteins and ligands, aiding drug discovery but requiring validation and complementary experimental data. Statins, including Atorvastatin, are commonly prescribed medications used to lower cholesterol levels and reduce the risk of cardiovascular diseases. Statins primarily work by inhibiting the enzyme HMG-CoA reductase, which is crucial for cholesterol production in the liver. This inhibition leads to decreased LDL ("bad") cholesterol and triglycerides while potentially increasing HDL ("good") cholesterol. Atorvastatin, in particular, not only lowers LDL and triglyceride levels but also offers additional benefits like improving endothelial function, stabilizing atherosclerotic plaques, and reducing oxidative stress and inflammation. These effects make Atorvastatin an effective treatment for preventing heart disease, strokes, and heart attacks. It is often used in combination with lifestyle changes such as diet, exercise, and weight loss. Beyond its lipid-lowering effects, Atorvastatin is used to treat conditions like familial hypercholesterolemia and is considered an essential therapy for individuals at high cardiovascular risk. Fluvastatin inhibits HMG-CoA reductase, reducing cholesterol production in the liver. This leads to increased LDL receptors, lowering LDL ("bad" cholesterol) in the bloodstream. It also helps increase HDL ("good" cholesterol) and slows plaque buildup in blood vessels. Fluvastatin is used to reduce heart disease risks and complications in people with coronary heart disease. It is suitable for adults and children aged 10 and above. It improves lipid profiles and reduces cardiovascular disease risk. Etoposide works by inhibiting DNA topoisomerase II, an enzyme that is crucial for DNA replication and repair. It blocks the re-ligation of DNA strands, causing DNA breaks that disrupt cell division. This results in apoptosis, particularly in rapidly dividing cancer cells. Etoposide is most effective during the S and G2 phases of the cell cycle. It primarily targets the topoisomerase II alpha isoform, essential for cell proliferation. However, its effect on the beta isoform may contribute to the risk of secondary cancers with long-term use.
Experimental
Aim
To predict the protein-ligand interactions and accelerating drug discovery by using molecular docking.
Objectives
• To prepare and optimize protein and ligand structures.
• To perform Molecular Docking Simulations
• To analyze and validate the results of binding interactions and scoring of the docking results.
MATERIALS AND METHODS
RESULTS AND DISCUSSION
The docking analysis assessed the interactions of Atorvastatin, Fluvastatin, and Etoposide with different target proteins, using Etoposide and its standard target protein (5NNE) as the benchmark. Etoposide exhibited a binding energy of -4.27 KJ/mol with 5NNE, indicating moderate binding stability. However, its high inhibition constant of 741.06 suggests weak inhibitory potential despite forming stable interactions. In comparison, Atorvastatin demonstrated a stronger binding affinity, particularly with 2IEJ (-5.76 KJ/mol) and 1LD8 (-5.72 KJ/mol), indicating a more stable interaction. The inhibition constant for Atorvastatin ranged from 59.56 to 384.13, significantly lower than that of Etoposide, suggesting enhanced inhibition efficiency. Fluvastatin, in contrast, exhibited weaker binding interactions, with binding energies ranging from -1.88 KJ/mol (6MBB) to - 4.57 KJ/mol (5UUP). The inhibition constants of Fluvastatin varied, with some values as low as 2.62 (6E3J), indicating poor inhibitory potential. Additionally, the van der Waals and hydrogen bonding interactions were strongest for Atorvastatin (-9.96 for 1LD8 and -9.02 for 2IEJ), followed by Etoposide (-6.62 for 5NNE) and Fluvastatin (ranging between -3.82 and - 6.82). Electrostatic interactions also favored Atorvastatin, with values as low as -1.22 (2IEJ), compared to 0.04 (5NNE) for Etoposide, suggesting that Atorvastatin forms more stable electrostatic interactions.
|
Pharmacokinetic Properties |
Atorvastatin |
Fluvastatin |
Etoposide |
|
Half-life (Hours) |
15-30 |
0.5-2.3 |
4-11 |
|
Bioavailability (%) |
12 |
19-29 |
IV-100 |
|
Protein binding (%) |
80-90 |
99 |
97-99 |
|
Solubility |
Lipophilic |
Lipophilic |
Amphiphilic |
|
Metabolism (cytochrome P450) |
CYP3A4 |
CYP2C9 |
CYP3A4 |
|
Urinary excretion (%) |
2 |
6 |
30-50 |
|
Faecal excretion (%) |
70 |
90 |
10-20 |
|
Common drug interaction (increase toxicity risk) |
Amiodarone Grapefruit Juice Protease inhibitors |
Diclofenac Amiodarone Protease inhibitors Azole antifungal |
Ketoconazole Ritonavir Clarithromycin |
Pharmacokinetics Of Statin
Atorvastatin
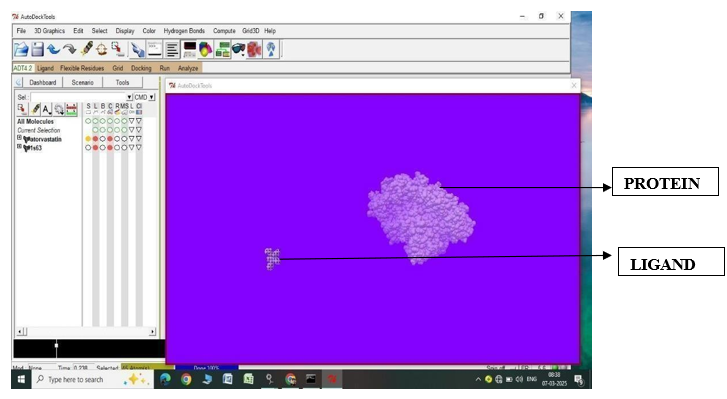
Fig.1.1: Ligand Atorvastatin and Protein 1S63 before In-silico docking process
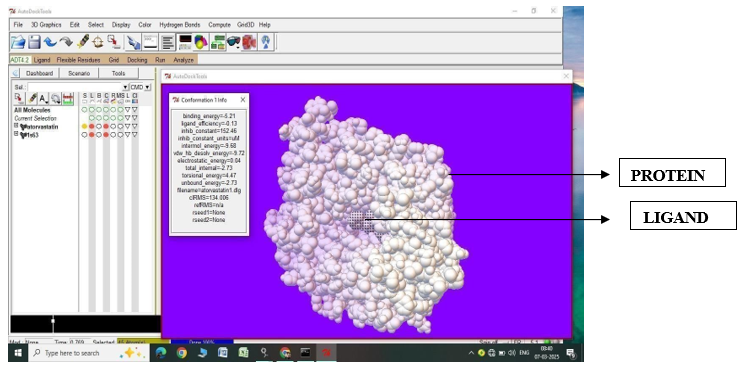
Fig.1.2: Ligand Atorvastatin and Protein 1S63 after In-silico docking process
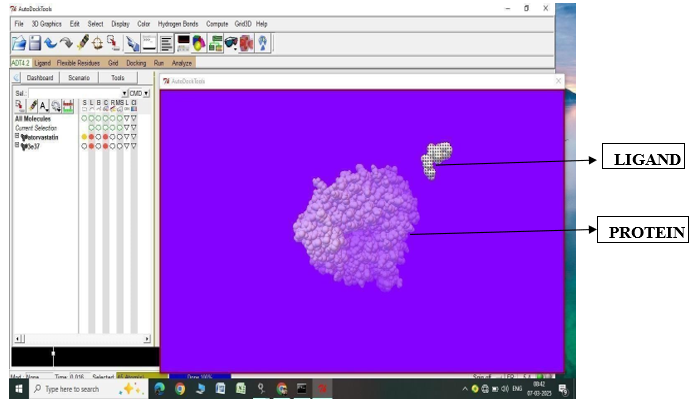
Fig.1.3: Ligand Atorvastatin and Protein 3E31 before In-silico docking process
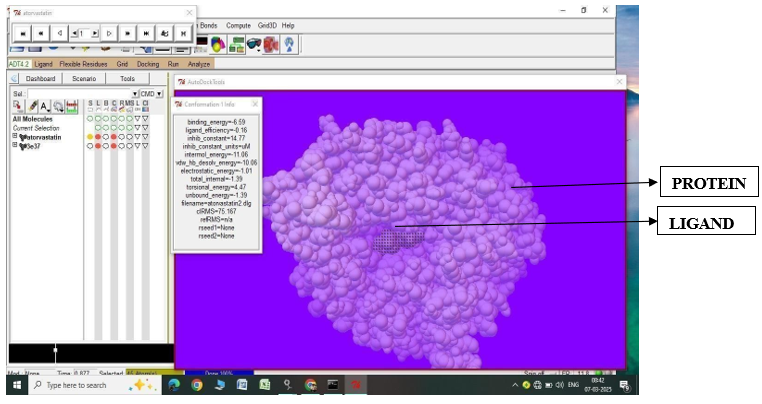
Fig.1.4: Ligand Atorvastatin and Protein 3E31 after In-silico docking process
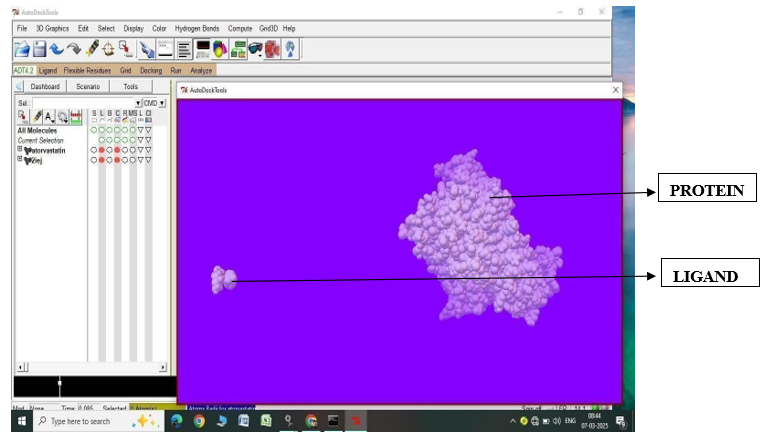
Fig.1.5: Ligand Atorvastatin and Protein 2IEJ before In-silico docking process
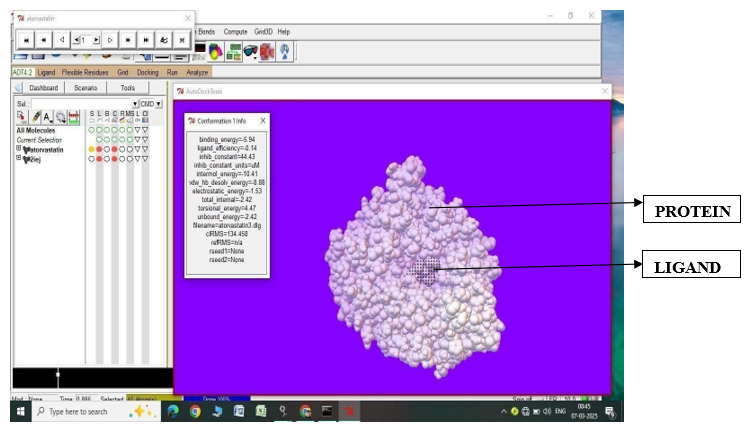
Fig.1.6: Ligand Atorvastatin and Protein 2IEJ after In-silico docking process

Fig.1.7: Ligand Atorvastatin and Protein 1LD8 before In-silico docking process
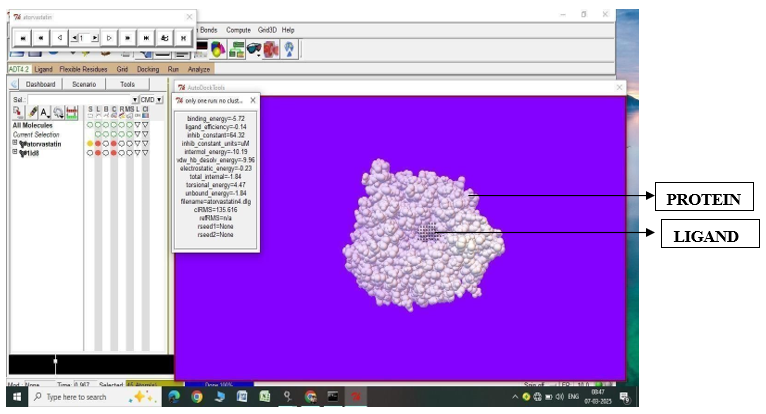
Fig.1.8: Ligand Atorvastatin and Protein 1LD8 after In-silico docking process
Fig.1.9: Ligand Atorvastatin and Protein 2H6H before In-silico docking process
Fig.1.10: Ligand Atorvastatin and Protein 2H6H after In-silico docking process
Fluvastatin
Fig.2.1: Ligand Fluvastatin and Protein 6MBB before In-silico docking process

Fig.2.2: Ligand Fluvastatin and Protein 6MBB after In-silico docking process
Fig.2.3: Ligand Fluvastatin and Protein 6E3I before In-silico docking process
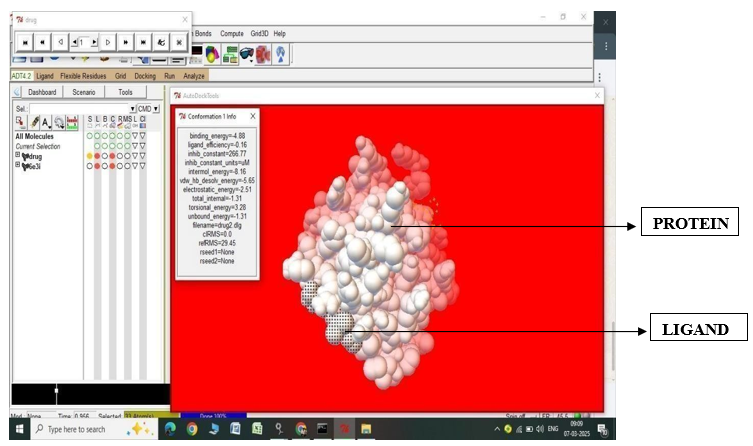
Fig.2.4: Ligand Fluvastatin and Protein 6E3I after In-silico docking process
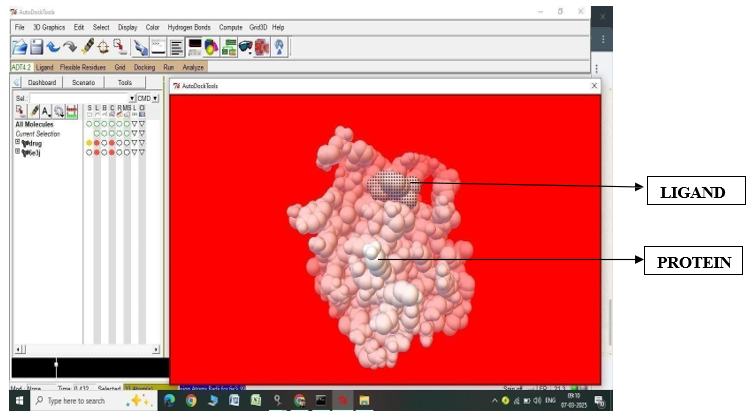
Fig.2.5: Ligand Fluvastatin and Protein 6E3J before In-silico docking process
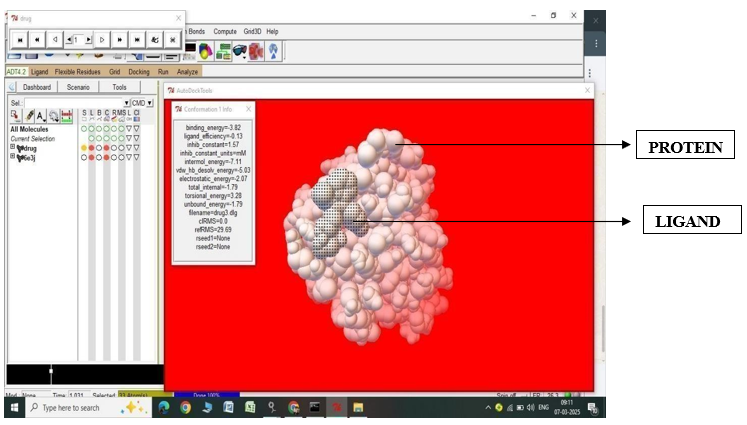
Fig.2.6: Ligand Fluvastatin and Protein 6E3J after In-silico docking process

Fig.2.7: Ligand Fluvastatin and Protein 5UUP before In-silico docking process
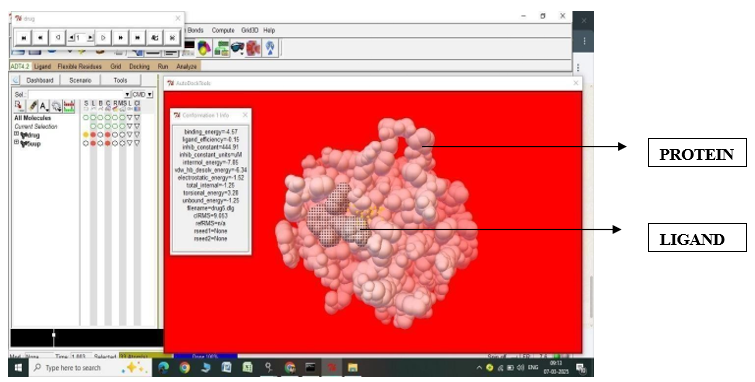
Fig.2.8: Ligand Fluvastatin and Protein 5UUP after In-silico docking process
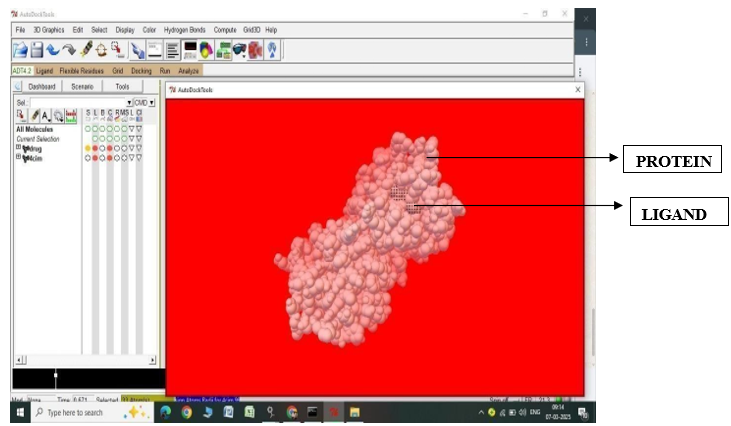
Fig.2.9: Ligand Fluvastatin and Protein 4CIM before In-silico docking process
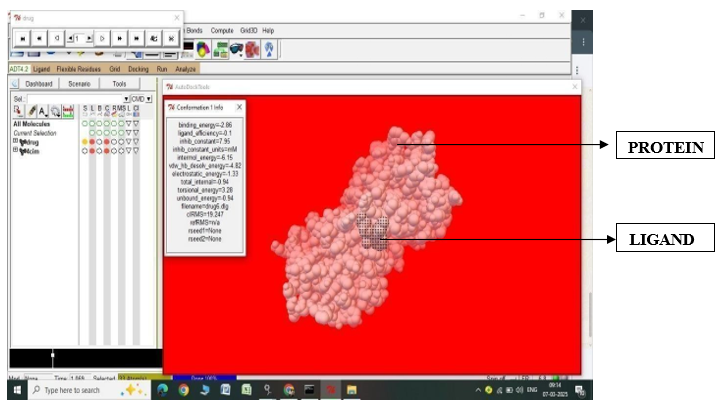
Fig.2.10: Ligand Fluvastatin and Protein 4CIM after In-silico docking process
Etoposide:
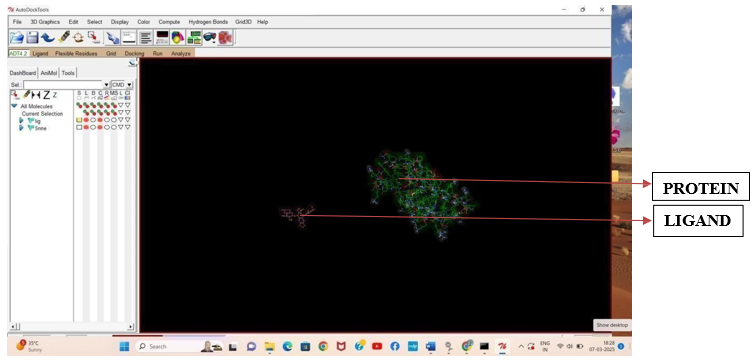
Fig.3.1: Ligand Etoposide and Protein 5NNE before In-silico docking process
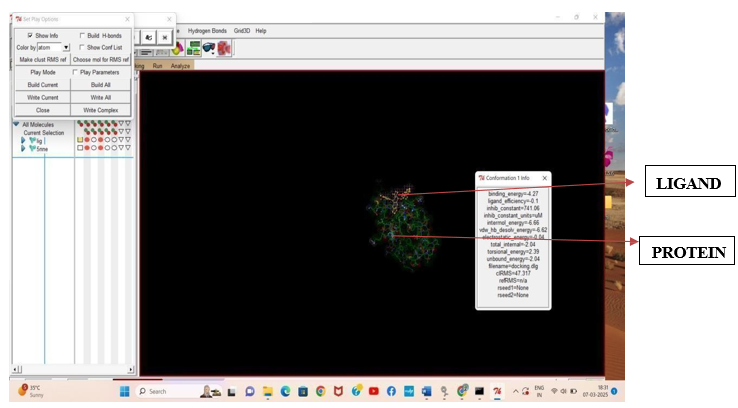
Fig.3.2: Ligand Etoposide and Protein 5NNE after In-silico docking process
Results of Protein - Ligand Interaction Using Auto dock
|
S. No. |
Compound name |
Protein |
Binding Energy (KJmol —1) |
Inhibition constant |
Vdw, h- bonding and solubility interaction |
Electro - static energy (kj Mol-1) |
Torsional energy (kj mol-1) |
|
1. |
Atorvastatin |
1S63 |
-4.96 |
230.78 |
-7.75 |
-1.69 |
4.47 |
|
2. |
Atorvastatin |
3E37 |
-5.18 |
159.13 |
-8.99 |
-0.67 |
4.47 |
|
3. |
Atorvastatin |
2IEJ |
-5.76 |
59.56 |
-9.02 |
-1.22 |
4.47 |
|
4. |
Atorvastatin |
1LD8 |
-5.72 |
64.32 |
- 9.96 |
-0.23 |
4.47 |
|
5. |
Atorvastatin |
2H6H |
-0.57 |
384.13 |
- 4.74 |
-0.3 |
4.47 |
|
6. |
Fluvastatin |
6MBB |
-1.88 |
42.23 |
-3.82 |
-1.34 |
3.28 |
|
7. |
Fluvastatin |
6E3I |
-2.74 |
9.84 |
-5.94 |
-0.08 |
3.28 |
|
8. |
Fluvastatin |
6E3J |
-3.52 |
2.62 |
-6.82 |
0.01 |
3.28 |
|
9. |
Fluvastatin |
5UUP |
-4.57 |
444.91 |
-6.34 |
-1.52 |
3.28 |
|
10. |
Fluvastatin |
4CIM |
-2.1 |
28.75 |
-5.82 |
0.44 |
3.28 |
|
11. |
Etoposide (Standard drug) |
5NNE |
-4.27 |
741.06 |
-6.62 |
0.04 |
2.39 |
|
S. No. |
Compound Structure |
Molecular Weight |
milogp |
TPSA |
No. of atoms |
No. of HBA |
No. of HBD |
No. of Rot .Bond |
|
1. |
Atorvastatin
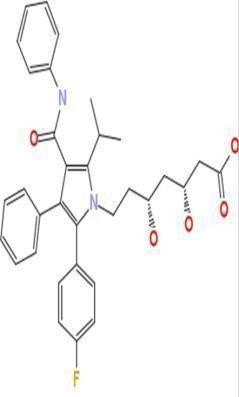
|
558.65 |
5.34 |
111.79 |
41 |
7 |
4 |
12 |
|
2. |
Fluvastatin
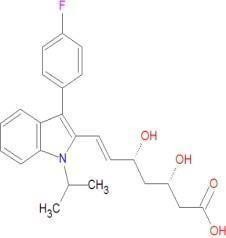
|
411.47 |
4.14 |
82.69 |
30 |
5 |
3 |
8 |
|
3. |
Etoposide (Standard drug)
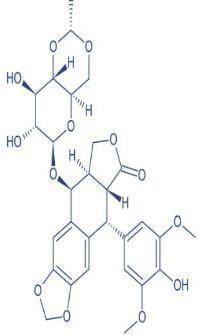
|
588.56 |
1.4 |
311.2 |
32 |
13 |
4 |
9 |
Molecular Properties of Drug Compound
DISCUSSION
The docking study highlights that Atorvastatin exhibits stronger and more stable interactions with specific target proteins compared to Etoposide, which is an already established chemotherapeutic drug. The lower binding energy and inhibition constant of Atorvastatin suggest that it may function as a more potent inhibitor against certain proteins. Notably, proteins 2IEJ and 1LD8, which showed the highest affinity for Atorvastatin, are associated with enzymatic pathways involved in cellular metabolism and proliferation. Strong interactions with these proteins suggest that Atorvastatin could influence crucial biological pathways, potentially leading to alternative therapeutic uses beyond its conventional role in cholesterol reduction. Etoposide’s standard target protein, 5NNE, is well-documented in cancer research and is involved in DNA topoisomerase inhibition, which is essential for halting tumor cell replication. However, the docking results indicate that Atorvastatin’s interactions with 2IEJ and 1LD8 are stronger than Etoposide’s interaction with 5NNE, suggesting that Atorvastatin could be a promising candidate for drug repurposing, particularly in pathways related to apoptosis or cell proliferation regulation. Several studies have explored statins like Atorvastatin for their potential anticancer properties. According to research by Zaky MY et al. (2023), statins exhibit pro-apoptotic and anti-proliferative effects in various cancer cells, which aligns with the strong binding observed in this study[40]. Another study by Juarez D et al. (2021) supports that Atorvastatin can downregulate key cancer-related proteins, including those involved in the mevalonate pathway, which is crucial for tumor growth. This suggests that Atorvastatin’s strong interaction with proteins like 2IEJ and 1LD8 might have biological relevance beyond lipid metabolism, reinforcing the idea that it could be repurposed for oncology treatments.[41] Additionally, the study by Dichtl W et al. (2003) discusses how Atorvastatin’s interaction with hydroxymethylglutaryl-CoA (HMG-CoA) reductase impacts inflammation and cell signaling, mechanisms also relevant in cancer progression. The fact that Atorvastatin showed stronger electrostatic interactions than Etoposide in this docking study further supports its potential as a multifunctional drug that can engage with various biological targets.[42] 44 In contrast, Fluvastatin exhibited the weakest interactions among the three drugs, indicating that its potential for repurposing in alternative therapeutic areas may be limited. Its higher binding energy and lower inhibition efficiency suggest that it is less effective in forming stable protein-ligand complexes. However, prior research Okubo K et al., (2020) has indicated that Fluvastatin might still possess moderate anticancer activity, though it appears to be less potent than Atorvastatin in targeting crucial cellular proteins.[43] From a pharmacological perspective, Etoposide remains the gold standard in targeting DNA topoisomerases for cancer therapy, but its higher inhibition constant and weaker electrostatic interactions suggest that it may not be the most efficient option for targeting proteins beyond its conventional application. Meanwhile, Atorvastatin’s ability to form stronger van der Waals and hydrogen bonding interactions with multiple proteins raises the possibility that it could have broader therapeutic implications, including in oncology and metabolic diseases. These findings suggest that further molecular dynamics simulations and wet- lab validation are necessary to confirm Atorvastatin’s potential as a repurposed drug. Exploring its impact on cancer pathways and metabolic processes in experimental settings will help determine its suitability for clinical applications beyond its traditional use in cardiovascular diseases.
CONCLUSION
Molecular docking has emerged as a vital tool in drug discovery, enabling researchers to predict protein-ligand interactions and design novel therapeutics. With its applications in virtual screening, lead optimization, and structure-based drug design, molecular docking has revolutionized the field of pharmaceutical research. As computational power and algorithmic sophistication continue to advance, molecular docking is poised to play an increasingly important role in the discovery of new medicines. Atrovastatin and Fluvastatin exhibit varying degrees of binding affinity to different protein targets, with Atrovastatin generally showing stronger binding energies. The results suggest that Atrovastatin may be a more potent inhibitor than Fluvastatin, and provide valuable insights for further optimization and development of these compounds as potential therapeutic agents.This study indicates that Atorvastatin exhibits stronger binding interactions and inhibitory potential compared to Etoposide, suggesting that it may be a promising candidate for repurposing in targeted therapies. The lower binding energy, enhanced inhibition potential, and stable interaction forces support Atorvastatin’s effectiveness in protein- ligand binding. Although Etoposide remains an established drug with known therapeutic efficacy, its weaker binding interactions and high inhibition constant suggest that alternative drugs should be explored for better interaction stability. Fluvastatin, on the other hand, displayed the weakest interactions and is the least promising among the tested compounds. Further molecular dynamics simulations and in vitro studies are required to validate Atorvastatin’s potential as a stronger alternative to Etoposide for specific protein targets. It is necessary to carry out further studies in order to clarify the anticancer mechanism and establish that the bioactive molecules are in nano-form in order to improve the bioavailability and effectiveness of the drug. The overall, the research finding exposes that, most of the synthesized compounds were active towards all the screened activities.
REFERENCES






Soundariya M.*, K. Premavathi, Arivumathi R., Nandhitha G., Parthiban K.V., Sandhiya S., Molecular Docking: A Computational Approach to Predict Protein-Ligand Interactions and Accelerating Drug Discovery, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 684-703 https://doi.org/10.5281/zenodo.15340579
 10.5281/zenodo.15340579
10.5281/zenodo.15340579
