1 Gujarat Technological University, Ahmedabad, Gujarat, India cum, Assistant Professor, Department of Pharmaceutics and Pharmaceutical Technology, L M College of Pharmacy, Ahmedabad - 380009, Gujarat, India.
2Shree S K Patel College of Pharmaceutical Education & Research, Ganpat University, Mehsana-984012, Gujarat, India.
A significant hurdle in modern drug development is the deficient aqueous solubility of approximately 40% of new small molecules, which restrict their oral absorption and bioavailability, a problem inherent to BCS Class II and IV drugs. Self-Microemulsifying Drug Delivery Systems (SMEDDS) have emerged as a highly competent lipid-based strategy to overcome this challenge. These formulations are isotropic mixtures of oil, surfactant, and a co-surfactant that spontaneously form a fine oil-in-water microemulsion upon gentle agitation in aqueous gastrointestinal fluids. This self-emulsification process, driven by extremely low interfacial free energy, creates thermodynamically stable droplets, typically under 250 nm, that keep the drug dissolved and enhance its intestinal penetration. The construction of a robust SMEDDS formulation is a systematic process, beginning with solubility screening to select optimal excipients. A pseudo-ternary phase diagram is then constructed to map the specific component ratios that yield a stable microemulsion region, directing the final formulation choice. While offering key advantages like improved stability and potentially reduced food effects , SMEDDS present hurdles such as potential GI irritation from high surfactant loads and drug precipitation upon dilution. To lessen leakage and stability issues with liquid forms, they can be solidified onto inert carriers. Rigorous characterization of the final product for droplet size, zeta potential, and in vitro drug release ensures its quality and performance, solidifying SMEDDS as a fundamental platform for converting poorly soluble drugs into viable oral therapies.
The reality that oral formulations make up about 80% of marketed medications reveal their patient-friendly nature, good safety record, and affordability(1). Creating oral preparations for BCS Class II and Class IV drug candidates presents many setbacks for formulation scientists. It is sometimes stated that approximately 40% of recently discovered small molecules have limited aqueous solubility, which limits their ability to dissolve when taken orally(2). Poor solubility leaves only a small fraction in solution, resulting in slow, variable, and frequently incomplete absorption—a problem integral to Class II (low solubility) and further compounded in Class IV (low solubility and low permeability)(3). A drug must first dissolve in gastrointestinal fluids and then penetrate the intestinal mucosa for effective absorption(4). A poorly soluble drug's absorption and bioavailability can be enhanced by using the appropriate formulation. Specifically, lipid-based formulations, which are composed of oils, surfactants, and co-surfactants, aid in maintaining the drug's solubility and enhance its absorption in the gastrointestinal tract (5). Since their initial report by Hoar and Schulman in 1943, microemulsions have found extensive use in everything from medicine solubilization to beverages and cosmetics. Microemulsions are thermodynamically stable, isotropic liquid systems made of water, lipids, and amphiphiles (usually a surfactant and a co-surfactant), as opposed to ordinary emulsions, which are kinetically unstable and ultimately phase-separate(6). SMEDDS are gaining the most interest among the many lipid delivery methods, including liposomes, solid lipid nanoparticles, self-dispersing tablets, and solid solutions. Compared to many alternatives, they are simpler to create and scale up, stable, and self-dispersing (7)(8).
LIPID-BASED DRUG DELIVERY
BCS Class II (low solubility, high permeability) and BCS Class IV (low solubility, low permeability) comprise a significant fraction of recently identified small compounds. By retaining the medication soluble during gastrointestinal transit and fostering a lipophilic milieu that inspires partitioning into the intestinal absorptive epithelium, lipid-based drug delivery systems (LBDDS) improve absorption (9) (10). Solid lipid nanoparticles (SLN), liposomes, lipoplexes, macroemulsions (coarse emulsions), microemulsions, and self-microemulsifying drug delivery systems (SMEDDS) are examples of lipid-based formulations. Lipid platforms have seen a significant increase in interest in recent years, with SMEDDS receiving special attention(11).
In 2000, Pouton proposed a widely adopted classification of lipid-based drug delivery systems, summarized in the table below (12).
Table 1 Classification of lipid-based drug delivery systems
|
Parameter |
Type I |
Type II |
Type III |
Type IIIA |
Type IV |
|
Triglycerides / mixed glycerides |
100% |
40–80% |
40–80% |
20% |
0% |
|
Surfactants |
– |
20–60% (HLB < 12) |
20–40% (HLB > 11) |
20–50% (HLB > 11) |
30–100% |
|
Hydrophilic co-solvents |
– |
– |
0–40% |
20–50% |
0–50% |
|
Dispersion particle size |
Coarse |
100–250 nm |
100–250 nm |
50–100 nm |
< 50 nm |
|
Digestibility |
Crucial |
Not crucial but likely |
Not crucial (may occur) |
Not important |
Not important |
Based on their excipient composition and water dispersion properties, lipid-based formulations are classified as Type I, II, III, and IV in Pouton's classification (13). Type III systems (SMEDDS) are isotropic preconcentrates of oil, high-HLB surfactant(s), and co-solvent(s) that are transparent to somewhat opalescent and that, when diluted in water, create thin microemulsions (10). When Type IV systems are dispersed, they produce very tiny colloidal dispersions (usually <50 nm), which are similar in droplet size to microemulsion systems. Type IV systems are made up mostly of high-HLB surfactants with hydrophilic co-solvents and contain little to no oil (9) (10).
Type I
A coarse O/W emulsion with only low emulsifier levels, such as ~1% polysorbate or lecithin, may be used to dissolve the drug in triglycerides or mixed glycerides.
Depend on pancreatic lipase/colipase to break down the lipids, produce additional amphiphilic molecules, and move the medication into colloidal aqueous phases. They initially disperse poorly (14).
Type II (SEDDS)
Systems that self-emulsify are isotropic mixtures of oils and lipophilic surfactants that, when diluted, produce fine O/W emulsions.
> ~25% (w/w) surfactant is usually required for self-emulsification(15)(16). It is possible to slow emulsification and encourage viscous liquid-crystalline phases at the interface by pushing the surfactant above about 50–60% (depending on the substance) (14).
Type III (SMEDDS)
Oil + hydrophilic surfactant(s) (HLB > 12) + co-solvent(s) make up self-microemulsifying systems.
Subcategories:
Type IIIA: Less hydrophilic surfactant/co-solvent and more lipid.
Type IIIB: Lower lipid, higher hydrophilic surfactant/co-solvent; disperses quickly to tiny droplets, but has a higher chance of drug precipitation upon dilution (14).
Type IV systems
Consist solely of hydrophilic co-solvents and surfactants, and when diluted with water, they produce colloidal micellar dispersions (17).
MECHANISM OF DRUG ABSORPTION FROM LIPID-BASED FORMULATIONS
Dissolving fat globules into a coarse emulsion with a high interfacial area, enzymatically hydrolyzing acylglycerols (mainly triglycerides) at the oil–water interface, and dispersing the resultant products into absorbable forms are the three interconnected steps that make up the digestion of lipid and lipid-based formulations(18). Triglycerides (TG) are hydrolyzed in the stomach by lingual and gastric lipases from the salivary glands and gastric mucosa, which produce amphiphilic products that aid in the formation of a crude emulsion that is emptied into the duodenum. These lipases are most active at pH values between 3 and 6, and medium-chain triglycerides hydrolyze more quickly than long-chain triglycerides. Pancreatic lipase and bile salts, which are released by the pancreas and gallbladder, respectively, stimulate lipolysis to produce Fatty acid (FA) and monoglycerides (MG) after entering the small intestine. Bile salts adsorb to incoming DG and TG, stabilizing the emulsion and reducing droplet size. Since MG and FA are efficient emulsifiers and released FA further improves lipase binding at the droplet surface, the process becomes self-promoting when pancreatic lipase, an interfacial enzyme, acts on emulsified TG droplets to create primarily 2-monoglyceride (MG) and two FA. On the surfaces of deteriorating oil droplets, multilamellar, liquid-crystalline intermediate phases may form during continuous lipolysis (19).
SELF MICROEMULSIFYING DRUG DELIVERY SYSTEM
Self-microemulsifying systems are isotropic combinations of oil, a surfactant, and a co-surfactant that spontaneously form a fine oil-in-water (O/W) emulsion when lightly stirred in gastric fluid or aqueous environments(20). In order to create SEDDS/SMEDDS, a variety of excipient combinations must be screened in order to find the best system that improves permeability, is physically stable, and guarantees efficient drug delivery (21).
Mechanism of Self-Emulsification
The system's interfacial free energy, which can be written as ΔG = Σ? πr²σ, where ΔG is the free energy, n is the droplet index, r is the droplet radius, and σ is the interfacial tension, controls the generation of self-emulsifying microemulsions. A lower interfacial tension lowers ΔG, which improves the emulsion's properties. When the mechanical energy necessary for dispersion is more than the energy required to form a new droplet surface, self-emulsification takes place. As it is expensive to create a new oil–water interface, conventional emulsions have a large free energy and can become unstable and phase-separate. SMEDDS, on the other hand, produce droplets nearly immediately because of a flexible interface that makes the system's free energy extremely low, sometimes even negative. An interface is first produced when water is gently stirred into an oil/surfactant/co-surfactant combination. Water then passes through this interface and dissolves into the oil phase up to its solubilization limit. A scattered liquid-crystalline phase develops when water absorption rises, and the degree of this phase depends on the concentration of surfactants. Fine droplets are created and the contact is disrupted by rapid water penetration. Regardless of their thermodynamic stability, microemulsions are dynamic because the material continuously switches between phases through processes like the fusing of tiny droplets, the fission of larger ones into smaller ones, and the fragmentation of droplets that subsequently coagulate with other droplets (7).
In vivo, microemulsions can be produced by the stomach and intestines' physiological movement. Self-microemulsifying drug delivery systems (SMEDDS) are commonly used to increase the oral absorption and solubility of poorly water-soluble medications. They create transparent microemulsions with oil droplets that are usually 100–250 nm in diameter. As the formulation crosses through the gastrointestinal tract in these systems, the medication stays dissolved within tiny oil droplets. Low drug-loading capacity, the requirement for high surfactant levels that could irritate the GI mucosa, temperature-sensitive storage, leakage from capsule shells, and general stability issues are some of the constraints of liquid SMEDDS. Liquid SMEDDS can be fixed onto inert carriers or solidified using methods like melt extrusion, melt granulation, adsorption onto porous excipients, or spray drying to get around these problems and create solid dosage forms. The resultant solid SMEDDS provide superior stability, simpler production, precise dosing, and increased patient compliance. They can be purchased as self-emulsifying controlled-release tablets, pellets, granules, microspheres, or minicapsules(22)(23).
SMEDDS, or self-microemulsifying drug delivery systems, are frequently utilized to address the low oral absorption and low solubility of water-insoluble medications. They naturally create oil-in-water microemulsions in the gastrointestinal tract, which facilitate rapid medication dissolution and improve intestinal epithelial penetration. Additionally, by entering the lymphatic route, the resultant microemulsion can lessen first-pass hepatic metabolism and increase bioavailability(24). The limited drug-loading capacity, the requirement for high surfactant levels (which may cause GI irritation), temperature-sensitive storage, probable capsule leakage, and general stability issues are some of the development challenges that SMEDDS face despite these benefits (25).
The distribution of the dissolved drug from the oil droplets into the aqueous dissolving medium is the rate-limiting step for drug dissolution in SMEDDS, and it is primarily controlled by the surfactant and co-surfactant concentrations. Functionally, SMEDDS and SEDDS create different dispersions: SMEDDS form clear microemulsions with mean droplet sizes usually below ~250 nm, while SEDDS commonly generate opaque emulsions with mean droplet sizes over ~300 nm (8). The type/HLB and concentration of the surfactant, the compatibility of the oil-surfactant pair and the selected oil:surfactant (or oil:Smix) ratio, and the physiological conditions during dispersion—particularly pH and temperature (and, in practice, factors like ionic strength and bile salts)—all affect self-emulsification (26).
Advantages of SMEDDS over other emulsions
One of the biggest headaches for drug developers is creating a pill that works, only to find the medicine inside won't dissolve properly in the gut, making it largely ineffective. This is where a clever formulation strategy called a Self-Micro emulsifying Drug Delivery System, or SMEDDS, comes to the rescue. Think of it as a sophisticated delivery package for the drug, crafted from a precise blend of oil, a surfactant, and a co-surfactant. When this carefully prepared mixture is swallowed and hits the stomach's watery environment, something remarkable happens: it spontaneously breaks apart into a stable cloud of incredibly tiny, drug-filled droplets. This immediate transformation into a microemulsion is crucial because it keeps the medicine fully dissolved and spreads it over a vast surface area, making it much easier for the body to absorb(11). Crafting the perfect SMEDDS recipe involves meticulous lab work, including mapping out the ideal component ratios to ensure this rapid and stable dispersion occurs. By solving the fundamental problem of poor solubility, this technology is a game-changer, unlocking the full potential of many promising drugs and turning them into effective oral treatments (11).
Limitations of SMEDDS
It's a frustrating paradox in medicine when a potentially life-saving drug fails simply because it won't dissolve properly in the body after being swallowed. To overcome this, formulation scientists have engineered a clever solution known as a Self-Micro emulsifying Drug Delivery System, or SMEDDS. A carefully designed pre-concentrate of oil, a surfactant, and a co-surfactant that holds the poorly soluble medicine in a ready-to-go liquid state. The remarkable part happens inside the stomach, where the gentle churning is enough for this mixture to spontaneously disperse into a stable, transparent cloud of incredibly tiny, drug-filled droplets, forming a microemulsion. This isn't guesswork; scientists use a tool called a pseudo-ternary phase diagram, which acts like a map to identify the perfect ratio of ingredients that guarantees this rapid and effective dispersion. By creating this massive surface area of pre-dissolved medicine, the system dramatically enhances the drug's absorption through the gut wall, often boosting its overall effect and even allowing it to bypass initial metabolism by the liver. While challenges like potential gut irritation from surfactants or leakage from capsules exist, these can often be managed by cleverly converting the liquid into a more stable solid form. Ultimately, SMEDDS technology is a powerful tool that transforms promising but difficult molecules into effective oral therapies (11).
Equilibrium solubility determination
Equilibrium solubility analysis is performed to choose the best oil, surfactant, and co-surfactant: an excess amount of the medication is taken into each excipient, then swirled to distribute it. To achieve equilibrium, the mixtures are left at room temperature for roughly three days while shaking them occasionally. To sediment any remaining medication, centrifugation is performed on the supersaturated samples for ten minutes at 12,000 rpm; further taking the supernatant and passing it through a membrane filter, diluting it with a suitable solvent, and using a suitable, approved analytical method (such as UV-Vis or HPLC) to measure the medicine that has dissolved (27) (28).
Construction of pseudo ternary phase diagram
By mapping composition regions that self-emulsify, a pseudo-ternary phase diagram is created to choose appropriate oils, surfactants, and co-surfactants for SMEDDS (21)(22). It assists in determining the proportions of constituents that, when diluted and gently mixed, create stable microemulsions(29). Usually, the aqueous titration method or the spontaneous emulsification method are used to create the diagram(9).
By illustrating the place where compositions spontaneously form transparent, thermodynamically stable micro/nano-emulsions upon dilution, a pseudo-ternary phase diagram is a straightforward map that is used to choose the optimal oil, surfactant/co-surfactant (Smix), and water ratios for SEDDS(30). It is constructed either by spontaneous emulsification/dilution—prepare mixes, dilute with water, and confirm rapid self-emulsification and small droplet size (e.g., by DLS) or by aqueous titration—mix fixed oil:Smix ratios (e.g., 9:1→1:1), titrate with water, and note the clarity–turbidity border. Plotting of points on triangle coordinates shows that self-emulsifying compositions that are sturdy to GI dilution are indicated by clear, isotropic (monophasic) regions, whereas muddy, separating (biphasic) regions do not(31). The diagram then directs the choice of component levels, particularly the co-surfactant content, to provide stability, fine droplets, and rapid dispersion (6) (21).
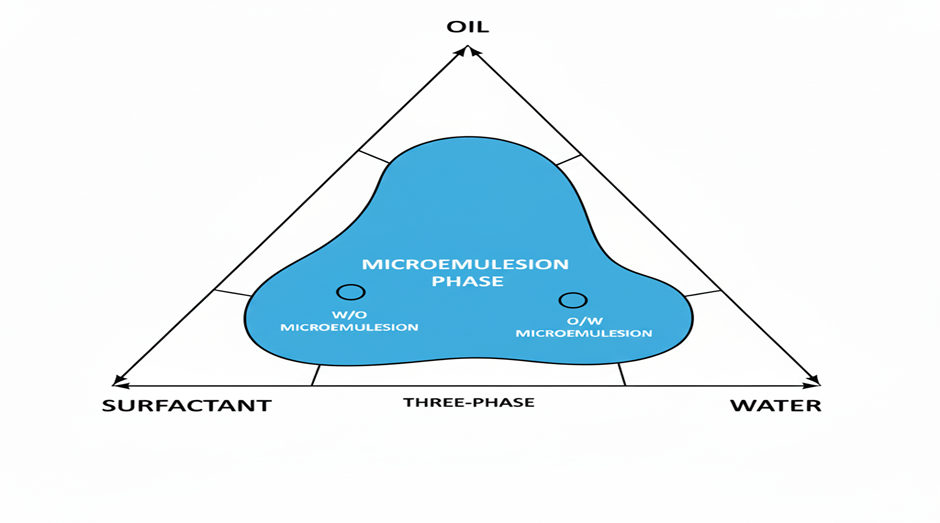
Figure 1 Pseudo ternary phase diagram
COMMONLY USED EXCIPIENTS FOR THE PREPARATION OF SMEDDS
Oil
Oral formulations can recruit a broad variety of natural, semi-synthetic, and synthetic lipids. Because they are frequently consumed, completely absorbed, and usually thought to be harmless, natural vegetable oils—which are combinations of triglycerides with varying chain lengths and degrees of unsaturation—make excellent base for SEDDS and SMEDDS(32). Long-chain triglycerides (LCT) make up the majority of vegetable oils, although medium-chain fats, particularly C12, are rich in coconut and palm kernel oils. Medium-chain triglycerides (MCT) from distilled coconut oil, primarily C8 (≈50–80%) and C10 (≈20–45%), are preferred in lipid systems because they often dissolve medications better (weight-based) than LCT and are less likely to oxidize. In addition to natural oils, medium-chain fatty acids and glycerides with hydrophilic groups are combined to generate a variety of liquid or thermo-softening (semi-solid) excipients that can be used as solvents, surfactants, wetting agents, emulsifiers, or co-emulsifiers for HPMC, soft-gel, or hard-gelatin capsules. Usually filled molten, thermo-softening excipients (m.p. ~26–70 °C) are practically restricted to hard-gelatin fills. Glyceryl monocaprylocaprate (Capmul MCM), glyceryl monostearate (GMS; e.g., Geleol/Imwitor 191), glyceryl distearate (Precirol ATO 5), glyceryl monooleate (Peceol), glyceryl monolinoleate (Maisine 35-1), glyceryl dibehenate (Compritol 888 ATO), and acetylated monoglycerides (Myvacet 9-45) are examples of common partial-glyceride excipients. Pharmacokinetically, LCTs (>C12) prefer intestinal lymphatic transport, whereas MCTs (C6–C12) are primarily delivered to the systemic circulation through the portal vein. MCTs are frequently utilized in lipid-based formulations due to their increased solvent capacity and oxidation resistance (11).
Table 2 Classification of glycerides for oral lipid-based formulations (SEDDS/SMEDDS) (17)
|
Class |
Examples |
Key characteristics (formulation-relevant) |
|
Triglycerides – Long-chain (LCT) |
Corn, soybean, olive, peanut, sesame, sunflower, castor oils, etc. |
GRAS; readily ingested/digested/absorbed. Lower self-dispersing ability and generally lower drug loading for intermediate log P drugs. After digestion, can offer higher solubilizing capacity. |
|
Triglycerides – Medium-chain (MCT) |
Fractionated coconut oil, palm-kernel oil; caprylic/capric triglycerides (e.g., Miglyol® 812, Captex® 355) |
Good solvent capacity for less-lipophilic drugs; good self-dispersion. Semi-synthetic MCTs are relatively oxidation-resistant. Widely used in SEDDS/SMEDDS. |
|
Mixed mono-, di- and tri-glycerides |
Imwitor® 988, Imwitor® 308, Maisine® 35-1, Peceol®, Plurol Oleique® CC49, Capryol®, Myrj® |
Amphiphilic; provide surface activity. Often replace conventional oils due to better self-dispersing behavior and higher solubilizing capacity for poorly water-soluble drugs; suitable for capsule filling. |
Surfactant
Choose surfactants for SEDDS that (i) provide a flexible interfacial layer that can speedily wrap and deform around droplets, (ii) lower oil–water interfacial tension to vastly low levels, and (iii) have the proper lipophilic–hydrophilic balance to provide o/w curvature. Safety and the lowest effective dose are critical since self-emulsifying formulae typically require between 30 and 60 percent w/w surfactant to create and sustain fine dispersions in the GI tract. However, higher amounts can irritate mucosa. LFCS Type III/IV systems usually use high-HLB, water-soluble surfactants (HLB ? 12) that form micelles over their CMC and encourage instantaneous o/w droplet formation and rapid spreading(33).
Typical choices include sorbitan/polysorbate pairings (Span 80 with Tween 80), polyglyceryl esters (e.g., Plurol Oleique CC 497), propylene-glycol esters (Capryol 90, Lauroglycol 90), PEG stearates (Mirj 45, Mirj 52), and PEG-15 hydroxystearate (Solutol HS 15). To increase oral bioavailability, these amphiphilic excipients function as wetting agents, emulsifiers/co-emulsifiers, and solubilizers (34).
Table 3 Surfactants classified by HLB (for SEDDS/SMEDDS) (34)
|
HLB band |
Type / family |
Examples (trade names) |
|
Low HLB (<10) |
Phosphatidylcholine & mixtures |
Phosphatidylcholine (incl. in propylene glycol/MCT, ethanol) |
|
Unsaturated polyglycolized glycerides (macrogolglycerides) |
Labrafil® M1944 CS, Labrafil® M2125 CS |
|
|
Sorbitan esters |
Capmul®, Capmul® S, Span® 20, Span® 40 |
|
|
Polyethoxylated alkyl ethers |
Brijs® 30, 52, 72 |
|
|
High HLB (>10) |
Polyoxyethylene sorbitan esters (polysorbates) |
Tween® 20, 40, 60, 80 |
|
Polyethoxylated fatty-acid esters |
Myrj® 52, Solutol® HS15 |
|
|
Polyethoxylated alkyl ethers |
Brijs® 35, 56, 78 |
|
|
Polyethoxylated glycerides |
Labrasol® (caprylo/caproyl macrogolglyceride) |
|
|
Polyoxyl castor-oil derivatives |
Cremophor® EL (polyoxyl 35 castor oil), Cremophor® RH40 (polyoxyl 40 hydrogenated castor oil) |
|
|
POE-POP block copolymers |
Poloxamer® 188, Poloxamer® 407 |
|
|
Saturated polyglycolized glycerides |
Gelucire® 44/14 (lauroyl), Gelucire® 50/13 (stearoyl) |
Favour high-HLB (≥12) surfactants (Tweens, Labrasol, Cremophor, Solutol, and Poloxamers) for quick o/w self-emulsification; they are frequently combined with a co-surfactant. Curvature and stability can be adjusted with the use of low-HLB materials (Spans, Labrafil). For GI tolerability and performance, aim for the lowest effective total surfactant (~30–60% w/w).
Co-surfactant
Seldom do single surfactants reduce the oil-water interfacial tension sufficiently or make the interfacial film sufficiently fluid to create microemulsions throughout a wide range(35). This is fixed by adding a co-surfactant; without it, the surfactant forms a rigid film and microemulsions only formed within a limited composition window; with it, the film becomes flexible, can take on various curvatures, and facilitates the creation of microemulsions over a larger range(36). Common co-surfactants are medium-chain alcohols (C3–C8), which also raise system entropy, increase interfacial fluidity, and decrease interfacial tension (34).
PREPARATION OF SMEDDS FORMULATION
To create a clear, transparent SMEDDS preconcentrate, combine the desired oil and surfactant:co-surfactant ratio (Smix), then add the prescribed dosage of medication and stir until the medicine dissolves fully(37).
Formulation of SMEDDS — Key Steps
VARIOUS CHARACTERIZATION OF SMEDDS FORMULATION
The dispersibility test
By using a USP dissolving apparatus II (paddle) to assess the oral SMEDDS's self-emulsification effectiveness. To produce mild agitation, keep 500 mL of filtered water at 37 ± 0.5 °C and set the paddle speed to 50 rpm. Allow the SMEDDS to disperse after injecting 1.0 mL of it into the medium. Using a predetermined dispersibility scale, score the in-vitro performance visually, noting the rate of emulsification, clarity/appearance, and any indication of phase separation. The formulation can then be categorized using a schematic decision flow according to its grade and the kind of dispersion that results from water dilution (24) (13).
Particle size
Droplet (particle) size is a direct yet effective predictor of success in SMEDDS: narrowly distributed, smaller droplets produce more surface area, which speeds up drug absorption and dissolution; they also produce clearer dispersions and higher self-emulsification grades. When growth or broad PDI develops, reformulation can be guided by supervising size and how it changes during stress tests (heating–cooling, centrifugation, and freeze–thaw) to identify metastability and approaching phase separation. In practice, a specified target size window makes it simple to create a quality control specification that guarantees reliable, stable, and bioavailable goods in each batch (42). The Coulter counter, photon correlation spectroscopy (dynamic light scattering), and microscopy are frequently used techniques to quantify droplet size. Because droplet diameter is a major indicator of the formulation's physical stability and regulates interfacial area, which in turn affects the rate and amount of drug release, precise sizing is crucial (43).
Zeta potential measurement
Electrophoretic light scattering (laser Doppler/ELS) is commonly used to determine droplet charge (zeta potential). Because free fatty acids at the oil–water interface deprotonate (–COO?) close to neutral pH, giving the surface a net negative charge, droplets in conventional SEDDS frequently exhibit a negative zeta potential(44). Physical stability is increased and coalescence is resisted thanks to this electrostatic repulsion. Keep in mind that bile salts, pH, ionic strength, and the type of surfactant or co-surfactant can all affect the charge's sign and magnitude; employing cationic surfactants can even cause the charge to reverse to positive (45).
Drug content
A predetermined quantity of the formulation is precisely dissolved in an appropriate solvent (such as methanol or acetonitrile: water), and then mixed or sonicated until it becomes clear(46). As necessary, dilute, filter (0.45 µm), and adjust the volume. The amount of drug present in the extract is determined using a validated analytical technique (such as HPLC or UV-Vis) in comparison to calibration standards made in the same solvent, and then the outcome is the percentage of drug content (label claim) (19).
Thermodynamic Stability Studies
Routine stability testing for SMEDDS/micro- or nanoemulsions is easier and less frequent than for coarse emulsions, although metastable systems—which appear to be fine but aren't actually thermodynamically stable—still need to be ruled out(47). To accomplish this, thermodynamic stability tests are performed, which usually involve centrifugation (e.g., 3,000–5,000 rpm, 15–30 min), heating–cooling cycles (e.g., 4↔40 °C), and freeze–thaw cycles (e.g., −20↔25 °C)(48). Unless chemical changes (oxidation, pH drift, etc.) are predicted to occur that could change components and destabilise the system, the formulation usually only requires limited ongoing tests throughout storage if it exhibits no phase separation and little change in droplet size/PDI following these stresses (19) (49).
Emulsification time
By putting the formulation in water and watching for spread, emulsification is evaluated. A milky look, no emulsion, or obvious oil droplets are scored as undesirable, while quick, even spreading was rated as acceptable. They are confirmed by spectrophotometric % transmittance (%T). The bigger droplet size, which scatters lighter and decreases %T, is the cause of the decreased transparency (49).
Self-Emulsification Time
USP Apparatus II (paddle) is used to assess self-emulsification effectiveness following thermodynamic stability screening. paddle is set to 50 rpm and equilibrate 500 mL of distilled water at 37 ± 0.5 °C. Emulsification is the process by which a uniform, clear/translucent dispersion forms without any discernible oil droplets or aggregates. 1.0 mL of the SMEDDS formulation is added dropwise into the medium, and it is noted how long it takes (49).
In vitro drug release study
To assess in-vitro drug release, a USP Dissolution Apparatus II (paddle) is used. To quantify drug release, each sample is placed in 900 mL of dissolution medium. At predefined intervals, aliquots is taken out, filtered (e.g., 0.45 μm) and subjected to analysis using a suitable validated method (e.g., UV–Vis or HPLC); the withdrawn volume is promptly replaced with fresh medium to maintain a constant volume (22).
Transmission Electron Microscopy
The morphology of the droplets is observed by using a transmission electron microscope (TEM) as a visual aid. Water is used to dilute(50). To examine the morphology of formulations, a drop of the diluted microemulsion is immediately placed on the holey film grid (51).
Viscosity
Using a jacketed cup with temperature control, viscosity is determined using a Brookfield viscometer. After loading and equilibrating around 10 mL of the SMEDDS for about five minutes, a suitable spindle is operated stepwise at 30 to 200 rpm, taking dial readings at each speed to determine and report apparent viscosity (vs. shear rate) (52).
pH
To account for GI circumstances, the pH of SMEDDS/microemulsions should be determined both neat and along a specified aqueous-dilution series. Weak organic acid systems frequently exhibit a non-linear pH trend with dilution: after the aqueous phase becomes continuous and the acid is nearly completely in water, additional dilution primarily dilutes H?, causing the measured pH to rise slightly. At low to medium water fractions, pH can decrease as acids partition into the aqueous phase and ionize (more H? released). pH of the SMEDDS is measured after the appropriate dilution. And the values should be physiologically acceptable (52).
Stability study
Under ICH Q1A(R2), the final product in the market pack at long-term and accelerated conditions (add intermediate if accelerated shows change)is kept, sample on a fixed schedule, and test with stability-indicating methods for assay, degradants, dissolution/release, and key physical attributes to determine the stability of the product (49).
CONCLUSION
Self-microemulsifying drug delivery systems (SMEDDS) offer a robust, scalable strategy to overcome poor aqueous solubility and erratic oral absorption of BCS II/IV drugs. As isotropic mixes of oil, high-HLB surfactant(s) and co-surfactant/co-solvent, they disperse in GI fluids to form clear microemulsions (typically <250 nm) that keep drug in solution and increase interfacial area for rapid uptake. Rational development follows a clear path: solubility screening, pseudo-ternary phase mapping to locate self-emulsifying regions, and targeted characterization (dispersibility, droplet size/PDI, zeta potential, viscosity, pH, release). While advantages include improved bioavailability, potential lymphatic transport, and simple manufacture, key risks remain—GI irritation from high surfactant loads, dilution-induced precipitation, and capsule compatibility/leakage—mitigated by judicious excipient choice, supersaturable designs, and conversion to solid SMEDDS when needed. Overall, the evidence supports SMEDDS as a foundational platform for translating lipophilic candidates into reliable oral products.
FUTURE PROSPECTS
Next-generation SMEDDS should emphasize (i) patient-friendly solids (spray-dried/adsorbed or melt-processed S-SMEDDS) to curb leakage and enhance stability, (ii) supersaturable systems using polymers/precipitation inhibitors to reduce surfactant burden while maintaining high apparent solubility, (iii) biorelevant, digestion-linked in-vitro tools with IVIVC to predict performance and lymphatic routing, (iv) QbD/DoE and computational screening (mixture designs, ML-aided excipient selection) to shrink development cycles, (v) targeted absorption via judicious MCT/LCT use, permeability modulators, and bile-salt–aware designs, and (vi) regulatory-ready stability (ICH Q1A/B) with stability-indicating methods and oxidative controls for lipid excipients. Integration of these directions should yield lower-surfactant, scalable SMEDDS with consistent clinical exposure and clearer pathways to approval.
REFERENCES


Mitalkumari Patel, Praful Bharadia, A Self-Driven Approach to Deliver Lipophilic Drug Molecules: Self-Micro Emulsifying Drug Delivery System, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 2935-2949. https://doi.org/10.5281/zenodo.17985788
 10.5281/zenodo.17985788
10.5281/zenodo.17985788
