Bhagwan Mahavir University, BMEF Campus, Nr. Aakash E-space, Bharthana, Surat, 395017.
A unique and promising method for oral drug delivery is the Liquisolid technique, particularly useful for medications with limited or low aqueous solubility. This technique is especially effective for creating immediate-release and sustained-release formulations, as well as for highly permeable medications classified under BCS Class II. Currently, around 40 to 50 percent of medications on the market are water-insoluble, presenting challenges for the pharmaceutical industry in enhancing dosage forms' solubility and bioavailability. These challenges are effectively addressed through liquisolid compact technology. The inclusion of non-volatile solvents in the formulation ensures molecular dispersion, enhances medication wettability, and ultimately boosts solubility. This method increases the drug's surface area, improving both its bioavailability and aqueous solubility. To analyze this technique, pre-compression factors such as porosity, Carr's index, flow behavior, powder bed hydrophilicity, and saturation solubility are examined. The quality of the formulation is also monitored by assessing post-compression criteria, including homogeneity, weight variation, hardness, friability, wettability, and disintegration time. This technique incorporates coating materials like silica gel and disintegration agents, as well as carriers such as lactose, flour, and microcrystalline cellulose. The physicochemical properties of the formulation are characterized and evaluated using analytical techniques, including X-ray diffraction (XRD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and Fourier-transform infrared spectroscopy (FTIR).
The understanding of drug candidates regarding their physicochemical characteristics like crystal structures and salt formation and biological aspects like metabolizing enzymes and transporters had been gathered to a great extent with the emergence of combinatorial chemistry and novel high-throughput screening techniques. There have been many active pharmaceutical ingredients (APIs) manufactured as a consequence. Still, the majority of these drugs remain very lipophilic with poor water-solubility. It is estimated that around 40% of the newly marketed drugs and almost 60% of the designed chemical entities have solubility problems. Thus, improving the solubility and dissolution of these poorly water-soluble drugs, and enhance their bioavailability’s is a concern for many scientists working in the field of pharmaceuticals. (1) The bioavailability of these Biopharmaceutical Classification System Class II (BCS II) drugs is frequently restricted by their solubility and the rate of dissolution in the gastrointestinal Tract. Many different strategies have been developed to enhance the poor solubility of certain medications. Micronization, deleting the drug’s form and increasing the more common surface area, does not always work well, especially in hydrophobic drugs that tend to aggregate in the form of tablets or capsules. Also, while solid dispersion does enhance dissolution and has gained much attention recently, few products finishing it like Kaletra® and Gris-PEG® are available due to the topographical and structural instability marks it remains having as its main solid-stateimp. Another well-known approach, soft gel capsules, is too technologically advanced and costly. Other strategies like complexation, microencapsulation, or self-nanoemulsifyingroving ways, as well as solid-lipid-nanoparticles,(2)Have also been researched, but these other poorly soluble drugs, also give too low results.
The liquisolid technique is an innovative and effective approach for modifying the dissolution rates of water-insoluble medications. Introduced by Spires, this concept of drug delivery can enhance the dissolution rate of these challenging substances. Often referred to as “powdered solution technology,” it is utilized to convert water-insoluble drugs into solid dosage forms that release quickly. This process involves transforming a liquid into a dry powder that flows easily and can be compressed, achieved through the physical blending with specific carrier and coating materials.(5,6)
This method effectively converts a liquid drug solution into a free-flowing, dry-looking powder, which can improve the solubility, wettability, and bioavailability of poorly soluble drugs.(6,8)
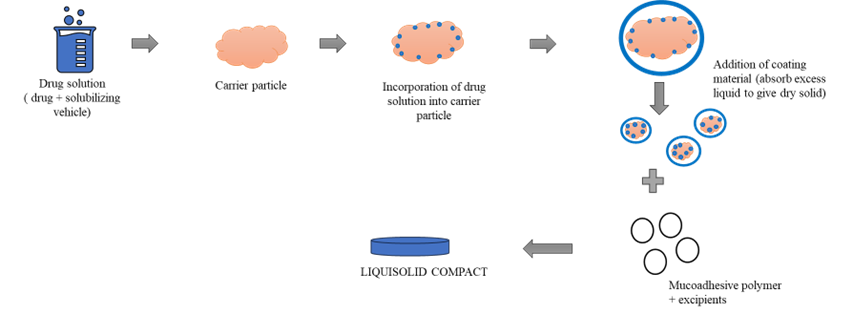
FIG 3.1; Formulation of liquisolid compact.(7)
Theories Of Liquisolid Compact
In the formulation of liquisolid systems, the precise quantities of powder excipients are determined using a mathematical model developed by Spireas. This model relies on the flowable liquid retention potential (Φ-value) and the compressible liquid retention potential (Ψ-number), which are specific constants for each powder-liquid combination.(9–11)
By utilizing these parameters, formulators can accurately calculate the necessary amounts of carrier and coating materials to achieve optimal flow and compression characteristics in liquisolid compact formulations. In the formulation of liquisolid systems, the liquid load factor (Lf) that ensures acceptable flowability (ΦLf) is determined by the equation:(13)
Lf=Φ+φ(1R)

Where:
R=Qq

This equation allows for the calculation of the maximum amount of liquid that can be incorporated into the liquisolid system while maintaining acceptable flowability. To achieve an acceptably flowing and compressible liquisolid system, the maximum liquid load on the carrier material mustn't exceed the value determined by the liquid load factor (Lf). This ensures that the formulation possesses the desired flow and compression properties necessary for effective processing and performance.(14) To quantitatively determine the necessary amounts of carrier (Q?) and coating (q?) materials required to convert a liquid formulation (W) into a powder with acceptable flowability and direct compressibility, the following equations are employed:(14)
Q0 = WL0
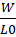
q0?=Q0R
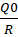
Here, L0 represents the liquid load factor, which is the maximum amount of liquid that can be incorporated per unit weight of the carrier material while still ensuring proper flow and compressibility. The parameter RRR is defined as the ratio of the weight of the carrier (Q) to that of the coating (q) material in the formulation. This mathematical approach facilitates the precise optimization of excipient proportions to achieve a liquisolid system with the desired processing and performance characteristics.
Table 2.1: Different Φ and φ values for different pharmaceutical excipients(15)
|
Sr No. |
Pharmaceutical Excipients |
Φ Value |
Φ Value |
||
|
|
|
propylene glycol |
PEG 400 |
propylene glycol |
PEG 400 |
|
|
Avicel PH102 |
0.16 |
0.005 |
0.224 |
0.242 |
|
|
Avicel PH200 |
0.26 |
0.02 |
0.209 |
0.232 |
|
|
Cab-o-Sil M5 with Avicel PH102 |
3.31 |
3.26 |
0.560 |
0.653 |
|
|
Cab-o-Sil M5 with Avicel PH200 |
2.56 |
2.44 |
0.712 |
0.717 |
Classification Of Liquisolid Compact
Liquisolid dosage is classified mainly on the following basis:
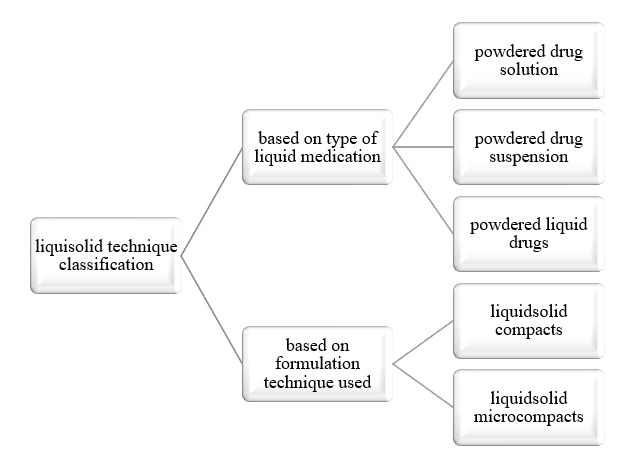
Based on the type of liquid medication
The first two may be produced from the conversion of drug solutions (e.g. prednisolone solution in propylene glycol) or drug suspensions (e.g. gemfibrozil suspension in Polysorbate 80), and the latter from the formulation of liquid drugs (e.g. clofibrate, valproic acid, liquid vitamins, etc.), into liquisolid systems.(15,16)
Liquisolid compacts are prepared using the previously outlined method to produce tablets or capsules, whereas the liquisolid microsystems are based on a new concept to produce an acceptably flowing admixture for encapsulations.(16,17)
Pharmaceutical compounds exhibiting aqueous solubilities below 0.1 mg/mL encounter significant solubilization challenges, while even those with solubilities under 10 mg/mL frequently manifest formulation difficulties due to suboptimal solubilization. It should be of BSC class II and IV. It should be lipophilic for example.(18)
• Digoxin – Cardiac glycoside
• Nifedipine – Anti-hypertensive
• Clofibrate - Antihyperlipidemic
• Nevirapine – Anti-viral
• Carbamazepine – Anti-epileptic
• Ibuprofen - NSAID
• Hydrochlorothiazide – Diuretics
• Lovastatin – hypertriglyceride
• Ivermectin – Anti-parasiytr
• Griseofuluvin – Anti-fungal
• Budesonide – Anti-asthmatic
• Praziquantel – Anti-helmented
• Trimethoprim – Antibiotic
4.2 Non-Volatile Solvent
In liquisolid systems, the selection of non-volatile solvents is crucial. These solvents should be safe for oral administration, water-miscible, chemically inert, and possess low viscosity. Non-volatile solvents act as wetting agents, enhancing the wettability of the drug particles. This improved wettability reduces the interfacial tension between the drug and dissolution medium, leading to a decrease in contact angle and an increase in the effective surface area of the drug. Consequently, the drug's dissolution rate is enhanced, facilitating better solubilization. (19,20)
Table 4.2.1: Types Of Non-Volatile Solvents Are Employed in Liquisolid Systems
|
S. No. |
Non-volatile solvent |
HLB value |
|
|
Propylene glycol |
2.5 |
|
|
Polyethylene glycol 200 monostearate |
8 |
|
|
Polyethylene glycol 400 monostearate |
11.5 |
|
|
Polysorbate 80 |
15 |
|
|
CapryolTM 90 |
5 |
4.3. Carrier
Carrier materials should exhibit porosity and robust absorption characteristics that facilitate effective liquid uptake. Both carrier and coating substances are capable of incorporating only limited quantities of liquid while preserving acceptable flow and compression properties; thus, an elevation in the moisture content of the carrier can adversely affect powder flowability. In liquisolid systems, carriers must possess a porous surface coupled with a high capacity for liquid absorption. The pivotal attributes of these carriers include their specific surface area and liquid absorption capacity, enabling them to integrate substantial amounts of liquid within their matrix.(21)
Table 4.3.1: Types of carrier material employed in liquisolid system
|
S. No. |
Carrier material |
Specific surface area [m2/g] |
|
|
Microcrystalline cellulose |
1.18 |
|
|
Lactose |
0.35 |
|
|
Sorbitol |
0.37 |
|
|
Starch |
0.6 |
|
|
Fujicalin |
40 |
|
|
neusilin |
300 |
4.4. Coating Material
In liquisolid systems, the coating material must consist of fine particles (ranging from 0.01 to 5 µm in diameter) with high adsorptive capacity. These characteristics enable the coating material to effectively envelop the wet carrier particles and adsorb excess liquid, thereby yielding a dry-appearing powder. Moreover, the coating material plays a critical role in ensuring complete surface coverage and preserving the overall flowability of the powder blend.(22)
Table 4.4.1: Types of coating material employed in liquisolid systems
|
S. NO. |
Coating material |
Composition |
Specific surface area [m2/g] |
|
|
Cab-O-Sil® M5-P |
Untreated fumed silica |
220 |
|
|
Syloid ® |
Amorphous silicon dioxide |
312 |
|
|
Aerosil®200 |
Hydrophilic fumed silica |
200 |
|
|
Neusilin® |
Amorphous aluminomagnesium metasilicate |
44-250 |
Additives
The disintegration process of solid dosage forms significantly impacts drug release. Sodium starch glycolate is the most commonly utilized disintegrant in the formulation of liquisolid tablets. Polyvinylpyrrolidone (PVP) serves as another advantageous additive due to its ability to incorporate a substantial amount of drug into liquisolid systems while reducing the overall tablet weight. Additionally, hydroxypropyl methylcellulose (HPMC) is employed as a release-modifying agent, often functioning to prolong drug release from liquisolid tablets. (23)
A fixed quantity of carrier material—optimally porous with adequate absorption capacity—is blended with an accurately measured volume of the prepared drug solution, suspension, or liquid formulation. This wetted mixture is subsequently combined with a precisely determined amount of coating material and mixed to yield a dry, non-adherent, free-flowing, and readily compressible powder. Excipients composed of fine, highly adsorptive particles are particularly effective in this process. Finally, liquisolid compacts are produced by incorporating various adjuvants, such as lubricants and super disintegrants, into the final liquisolid formulation prior to compression or encapsulation.(24)
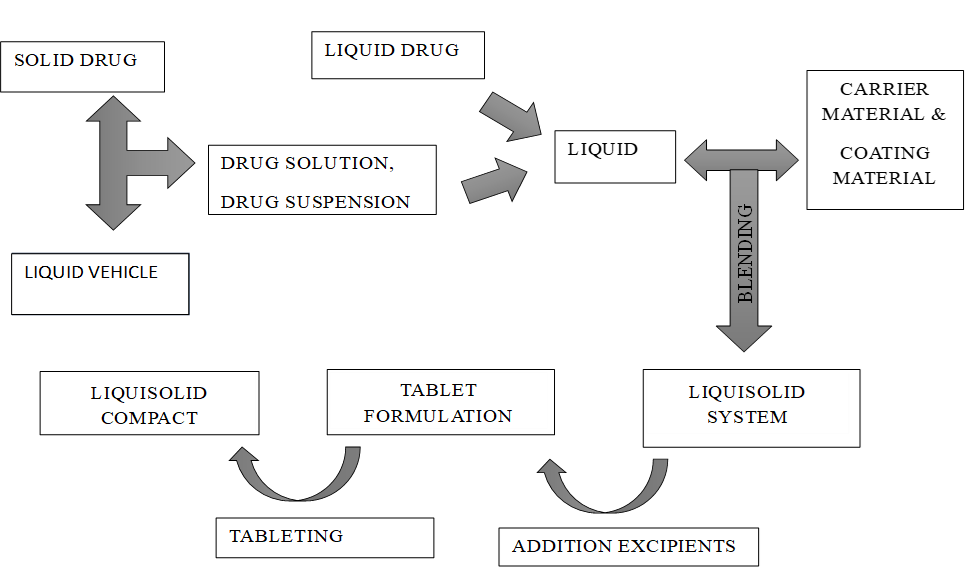
Figure 5.1: Method of preparation of liquisolid compact
Three main mechanisms are involved in enhancement of drug release from the liquisolid system are as follows.
The amount of surface area of the drug that can be delivered from a liquisolid system is significantly larger than that of the drug particles that are directly compressed. Tablets since the medication in the liquisolid system fully dissolves in the liquid medium and remains solubilized and molecularly disseminated in the powder substrate. As a result, the solubility limit improves with increasing drug content, increasing the percentage of medication that remains undissolved in the liquid vehicle and lowering the release rate. The fraction of the drug that is molecularly dispersed (FM) in the liquid-solid formulation directly correlates with the drug's release rate. Spireas defined FM as the ratio of the drug's actual concentration (Cd) in the liquid vehicle to its solubility (Sd).(25)
FM = Sd⁄Cd
Where FM = 1 Sd ≥ Cd
The liquisolid technique can enhance the drug's solubility. In order to enhance the drug's overall solubility in the aqueous dissolution medium, a liquisolid compact's tiny quantity of liquid vehicle is insufficient. A modest amount of liquid vehicle is enough to improve the poorly water-soluble drug's aqueous solubility if it functions as a co-solvent in a liquisolid system.(25)
By diminishing the interfacial tension between the tablet surface and the dissolution medium, the non-volatile solvent in the liquisolid system promotes drug particle wetting. As a result, the liquisolid system's contact angle is reduced in comparison to the conventional formulation, improving wettability. (25)
The flow behavior of liquisolid formulations is an important consideration in the development of solid dosage forms. As a result, evaluating the flow parameters of liquisolid powder mixes before compression is critical. This assessment can be carried out utilizing the following metrics
7.1.1. Angle Of Repose
The angle of repose is an important measure used to assess the flow parameters of the liquisolid powder combination prior to compression into tablets. Because liquisolid systems consist of liquid-loaded carriers and coatings, liquid quantity, excipient selection, and particle size all have a substantial impact on flowability.(24,26) The following calculation was used to determine the angle of repose (????):
tan θ = h/r
Where,
θ = angle of repose,
h = Height of the heap,
r = Radius of the heap.
7.1.2. Carr’s Compressibility Index
The main objective of Carr's compressibility index techniques is to ascertain the dry mixture's index of compressibility.
Carr's compressibility index may be computed using the formula below:(27)
Carr’s index = tap density-bulk density
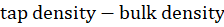
/ tap density X 100
7.2.3. Hausner’s Ratio
An essential characteristic for figuring out the flow characteristics of powders and granules is Hausner's ratio. The following formula can be used to compute this:(28)
Hausner’s ratio = Tapped density/Bulk density.
7.3.4. Fourier Transform Infrared Spectroscopy (FTIR)
FTIR analysis illustrates chemical interactions between the drug and excipients used in the formulation. The presence of peaks in a medication formulation without any extra peaks suggests no chemical interaction. (29)
The crystalline properties of a liquisolid compact combination are ascertained using XRD investigations. It is noted that in the liquisolid formulation, distinctive peaks vanish while additional carrier peaks remain. It shows that the medication changed into a stable or amorphous form.(30,31)
Differential Scanning Calorimetry (DSC) is used to determine how the excipients used in the preparation interact with one another. This can also be a sign of the stability studies' effectiveness. The medication is disseminated molecularly inside the system once it is in the form of a solution in a liquisolid formulation, as indicated by the total disappearance of distinctive peaks.(32)
Tablets are difficult to break during regular mobility and decompose appropriately after use. The mechanical strength is measured with it. The hardness of liquefied compact is measured using a Monsanto hardness tester and is expressed as kg/cm2.(33).
Since some extremely hard compressed tablets lose their cap section when they are abrasively damaged, hardness alone is insufficient to assess a tablet's strength. Friability is therefore the way to gauge the strength of the tablets. The friabilator is the device used to test a tablet's friability. After being weighed and placed in the friabilator, 20 tablets were allowed to revolve as per requirement. The tablets were taken out and weighed once again following the conclusion of the revolutions.(33,34)
The weight variation test is employed to determine if each tablet contains the appropriate dosage of medication. According to the IP 2022 guidelines, twenty tablets are randomly chosen and weighed. The average weight of these tablets is then calculated, ensuring that the weight of no more than two tablets differs from the average by more than 5%.(35–37)
A disintegration apparatus is used to measure a tablet's in-vitro disintegration time in compliance with Indian Pharmacopoeia (I.P.) specifications. A single tablet is put into each of the disintegration baskets' six tubes per the I.P. requirements. To provide consistent testing conditions, a perforated disc is inserted into every tube. In order to replicate gastrointestinal conditions, the device is run in a 1.2 pH buffer solution and kept at 37°C ± 2°C. Until the tablet disintegrates completely, the basket assembly experiences vertical oscillations at a frequency of 30 cycles per minute, which guarantees constant mechanical stress.(38)
Using 900 mL of dissolved media, a dissolution study was carried out over an hour using the USP Type 2 (paddle) method. The temperature was kept at 37?±?0.5 °C, and the device was adjusted to the necessary rotating speed. To keep the total volume constant, a 10 mL aliquot of the dissolution medium was taken out at prearranged intervals and replaced right away with an equivalent volume of fresh medium. A UV/Vis spectrophotometer was then used to evaluate the samples that had been collected.(39–42)
The purpose of the stability research is to determine the items' shelf life. The amount of time needed to lower the reactant concentration to 90% of its starting concentration is known as shelf life. The impact of storage on the drug release profile and the crushing strength of liquisolid compacts were examined in order to gather data on the stability of liquisolid systems. The hardness and drug release profile of liquisolid compacts may not be impacted by storage under various conditions, according to stability studies of liquisolid systems. This suggests that the technology is a viable method for increasing the release rate without causing issues with physical stability.(43)
CONCLUSION
To sum up, liquisolid compacts are a robust and innovative hybrid strategy for improving drug administration, especially for medications that are not highly soluble in water. The numerous advantages of liquisolid systems, such as better dissolving profiles, increased bioavailability, and the possibility of regulated release, have been emphasized in this review. A special platform that gets around the drawbacks of standard techniques is made possible by the combination of liquid medication formulations and conventional solid dosage form technology. Liquisolid compacts' hybrid nature provides major industrial benefits in terms of scalability and formulation flexibility in addition to making it easier to transform liquid pharmaceuticals into free-flowing, compressible powders. Developments in this area have broadened the variety of applications, from formulations with instant release to those with sustained release, showcasing the technique's adaptability and versatility. Future research into novel carriers, coating materials, and solvent systems is expected to improve and optimize liquisolid formulations. Furthermore, further investigation of process parameters and in vivo performance will be required to fully realize the therapeutic potential of this technology. Finally, liquisolid compacts show promise as a critical tool in the creation of next-generation drug delivery systems, opening the door to safer, more effective, and patient-friendly pharmacological therapies.
ACKNOWLEDGEMENT
I sincerely express my gratitude to Bhagwan Mahavir College Of Pharmacy for providing the necessary resources and support for this review article. I extend my heartfelt thanks to the esteemed Dean, for their encouragement and guidance. I also appreciate the invaluable assistance and insights from the faculty members whose expertise and support have greatly contributed to the completion of this work.
Conflict Of Interest
The authors declare that there are no conflicts of interest regarding the publication of this review article.
Funding Statement
The authors declare that no financial support was received for the research, authorship, and/or publication of this review article.
REFERENCES


Ekta Tiwari*, Dr. Velenti Chauhan, Liquisolid Compacts: A Hybrid Approach to Enhance Drug Delivery, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 3, 2895-2907 https://doi.org/10.5281/zenodo.15101657
 10.5281/zenodo.15101657
10.5281/zenodo.15101657
